Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Antoine Chaanine | + 3369 word(s) | 3369 | 2021-10-12 04:17:01 | | | |
| 2 | Peter Tang | -5 word(s) | 3364 | 2021-10-20 08:49:32 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Chaanine, A. Calcium Signaling and Mitochondrial Oxidative Capacity. Encyclopedia. Available online: https://encyclopedia.pub/entry/15159 (accessed on 28 June 2026).
Chaanine A. Calcium Signaling and Mitochondrial Oxidative Capacity. Encyclopedia. Available at: https://encyclopedia.pub/entry/15159. Accessed June 28, 2026.
Chaanine, Antoine. "Calcium Signaling and Mitochondrial Oxidative Capacity" Encyclopedia, https://encyclopedia.pub/entry/15159 (accessed June 28, 2026).
Chaanine, A. (2021, October 20). Calcium Signaling and Mitochondrial Oxidative Capacity. In Encyclopedia. https://encyclopedia.pub/entry/15159
Chaanine, Antoine. "Calcium Signaling and Mitochondrial Oxidative Capacity." Encyclopedia. Web. 20 October, 2021.
Copy Citation
The intermyofibrillar mitochondria constitute the majority of the three-mitochondrial subpopulations in the cardiac myocyte. They are also considered to be the most important in terms of their ability to participate in calcium and cellular signaling, which are critical for the regulation of mitochondrial function and adenosine triphosphate (ATP) production.
heart failure
mitochondria
metabolic remodeling
calcium
signaling
1. Introduction
The heart is an organ with high-energy demands necessary to maintain its continuous mechanical work [1]. The mitochondria, which are the powerhouse of the cell, are most abundant in the heart [2]. Their role extends beyond adenosine triphosphate (ATP) production to include ion homeostasis, participation in calcium signaling and redox balance, and are considered the hub of cell metabolism [3]. This multiplicity of mitochondrial (mt)-function is tightly coordinated to maintain cell survival, otherwise cell death ensues [4]. Perturbations in mt-homeostasis and metabolic remodeling constitute important pathophysiological processes in the development of heart failure (HF). To date, it is well known that fatty acid (FA) metabolism and mt-FA-β-oxidation are attenuated in compensated hypertrophy and progressively worsen upon progression to overt systolic heart failure (SHF) [1][5]. Glucose metabolism is initially up-regulated in compensated hypertrophy then is attenuated in progression to SHF [1][6]. Oxidative phosphorylation (OXPHOS) is attenuated at the onset of left ventricular systolic dysfunction and drop in ejection fraction, and is impaired further upon progression to overt SHF [7]. Molecular mechanisms and signal transduction pathways regulating mt-function and cardiac metabolism are partially known to date. The peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) and the peroxisome proliferator-activated receptor alpha (PPARα) signaling, which gradually decline in progression to overt SHF [8][9], are important players in the regulation of metabolism, primarily FA metabolism [10]. Protein kinase A (PKA) and its opponents protein phosphatase 1 (PP1) and calcineurin (PP2A) as well as the adenosine-monophosphate-activated protein kinase (AMPK) signaling, their interplay, play an important role in the regulation of cardiac metabolism and mt-function. Other kinases, such as protein kinase C epsilon (PKCε), calcium/calmodulin kinase II isoform delta (CaMKIIδ) and the Jun-N-terminal kinase (JNK), also modulate mt-function and dynamics. It is not known which of these pathways predominate and what is the sequence of derangements in signal transduction pathways that lead to the initiation and progression of HF. This derangement in signaling is also paralleled by derangements in calcium cycling [11] and calcium-mediated endoplasmic reticulum (ER)–mt crosstalk; thereby, leading to metabolic reprogramming and mt-dysfunction in a milieu of heightened oxidative stress that constitutes the hallmark of the syndrome of HF. The topology of the intermyofibrillar mitochondria in the heart places them at the hub of calcium signaling as they are neighbored, at a very close proximity, by the T-tubules, the longitudinal sarcoplasmic reticulum (SR) and the sarcomeres as we discuss below.
2. Mitochondrial Morphology, Topology and Function
Mitochondria are membrane bound organelles that are thought to have originated from symbiotic ancestors [12] and are found in almost all eukaryotic cells. They are comprised of an outer (OMM) and an inner mt-membrane (IMM). Between the OMM and IMM is the inter-membrane space (IMS) where the proton pump gradient forms across the IMM and is the sole determinant of mt-capacity for ATP production. The mt-matrix is surrounded by the IMM and is the site where most reactions related to metabolism and OXPHOS takes place. The mt-matrix also contains mt-DNA and the necessary mt-translation/elongation machinery that encodes 13 proteins that are essential for respiratory chain function in human [13][14]. However, the majority of mt-genes are encoded in the nucleus and are then imported into the mitochondria through a specialized mt-import and assembly systems [15].
There are three mt-subpopulations in the cardiac myocyte: the perinuclear, the intermyofibrillar and the subsarcolemmal mitochondria [16], Figure 1A. Of these, the intermyofibrillar mitochondria are most abundant and are most important due to their participation in calcium signaling and ATP production [17]. Each intermyofibrillar mitochondrion extends from one Z band to another and is surrounded by sarcomeres at each side and T-tubules at each end, Figure 1B. The longitudinal SR forms a network around the intermyofibrillar mitochondria and is tethered to them via tethering complexes [18], known as the ER-mitochondria encounter structure (ERMES) [15]. Thus, the OMM acts as a nodal membrane that connects the mitochondria with their surrounding and is the site where the majority of kinases and phosphatases are anchored to modulate mt-morphology and function and metabolism. Figure 2 is a schematic drawing summarizing the metabolic pathways, calcium cycling and implicated signaling regulating metabolism, calcium cycling and mt-function in the heart under normal physiological conditions.

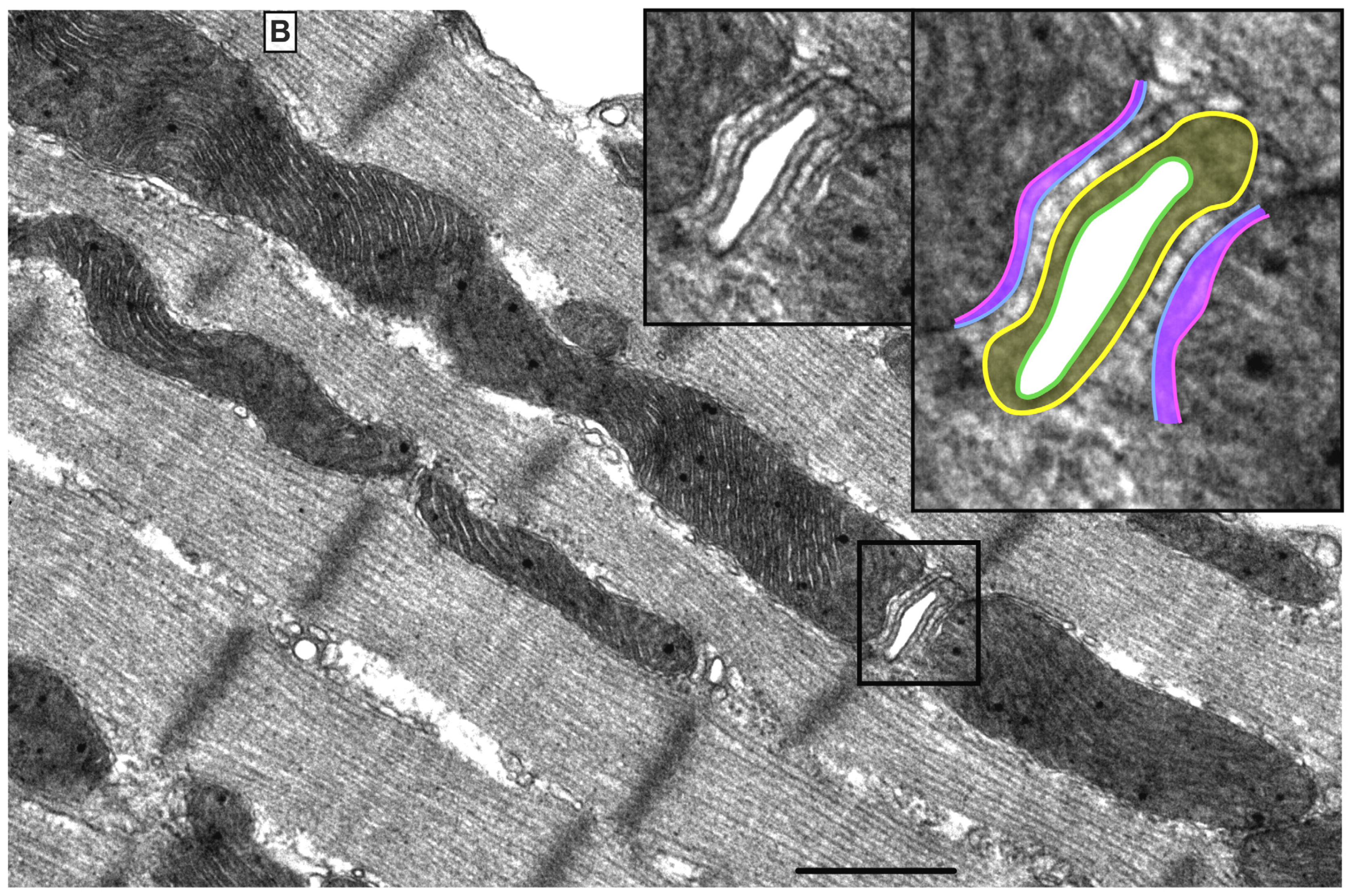
Figure 1. Ultrastructure of normal myocardium. (A) Transmission electron photomicrograph of adult cardiac myocyte showing perinuclear, intermyofibrillar and subsarcolemmal mitochondria. Red box in the figure is defining the zoomed image showing mitochondrion-containing autophagosome. Image 5k × magnified, scale bar 1 μm. (B) Transmission electron photomicrograph of adult cardiac myocyte showing the relationship of the intermyofibrillar mitochondria with the adjacent T-tubules and sarcomeres. Image 40k × magnified, scale bar 1 μm. Tracing in the zoomed image, defined by the black box, shows T-tubule (green line surrounding white area), junctional sarcoplasmic reticulum (yellow line with yellow shaded area), outer mitochondrial membrane (Blue line), inner mitochondrial membrane (pink line) and intermembrane space (purple shaded area).
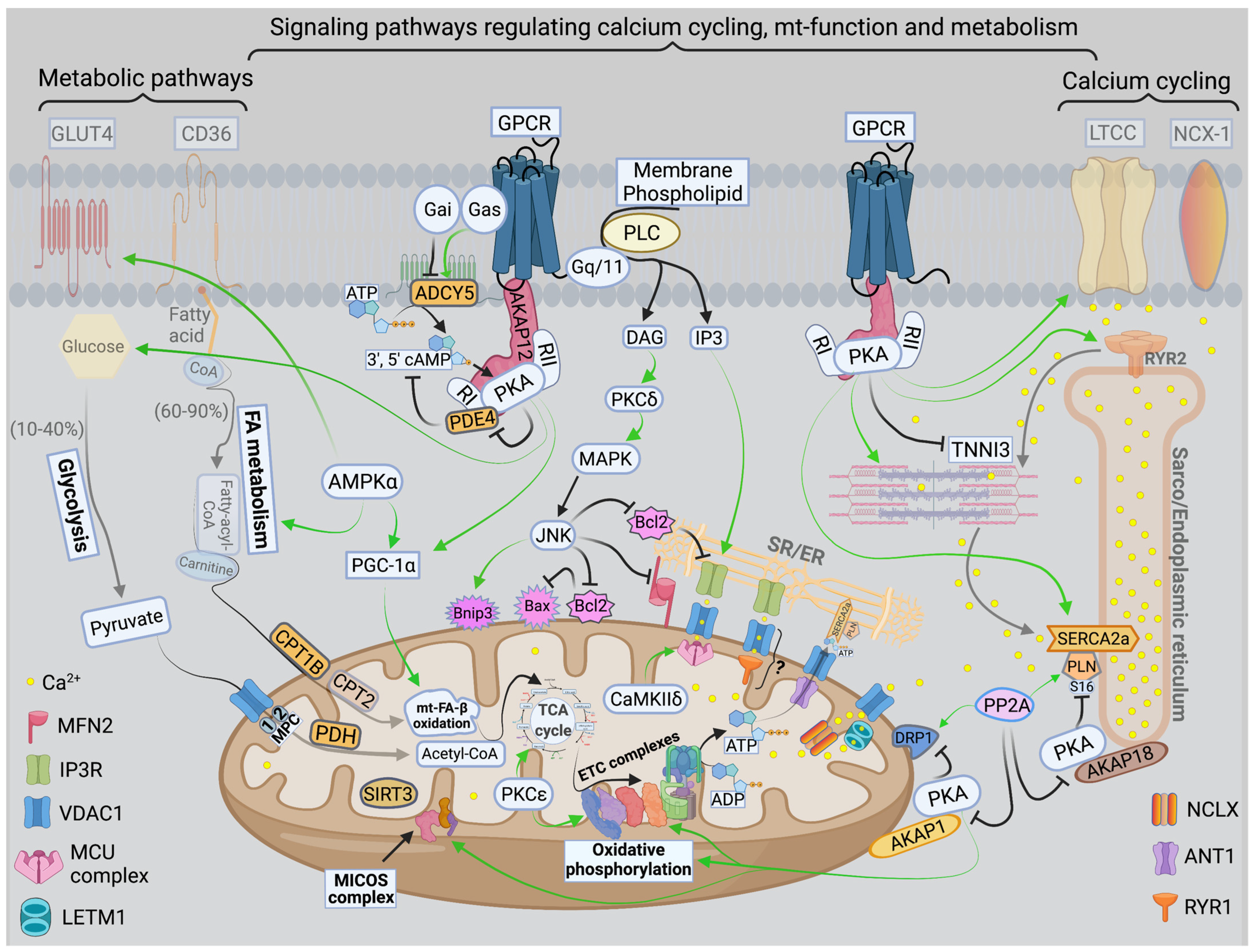
Figure 2. Schematic drawing showing metabolic pathways, calcium cycling and implicated signaling in normal myocardium. Green arrows promote signaling or activity. Metabolic pathways on the left show that in normal heart, fatty acid (FA) metabolism and mitochondrial (mt)-FA-β-oxidation contribute 60–90%, while glucose and pyruvate metabolism contribute 10–40% of the generated adenosine triphosphate ATP by oxidative phosphorylation (OXPHOS). On the far right is a presentation of calcium cycling and its regulation by protein kinase A (PKA) signaling. Please refer to text for details. In the center of the figure is a presentation of signaling pathways modulating mt-function and metabolism. The bottom center of the figure shows the tethering complexes that exist between the endoplasmic reticulum (ER) and neighboring mitochondria and how they crosstalk via calcium signaling. The presence of mt-calcium uniporter (MCU)-independent mt-calcium uptake via the ryanodine receptor 1 (RYR1) into the mt-matrix has not been clearly validated and is represented by a question mark (?). Abbreviations: GLUT4: glucose transporter member 4, CD36: FA transporter, MPC: mt-pyruvate carrier, PDH: pyruvate dehydrogenase, TCA: tricarboxylic acid, PKCε: protein kinase C epsilon isoform, PKCδ: protein kinase C delta isoform, CaMKIIδ: calcium/calmodulin kinase delta isoform, ADP: adenosine diphosphate, MFN2: mitofusin 2, IP3: inositol triphosphate, IP3R: inositol triphosphate receptor, VDAC1: voltage dependent anion channel 1, LETM1: leucine zipper and EF-hand containing transmembrane protein 1, NCLX: sodium/calcium/lithium exchanger, ANT1: ADP/ATP translocase 1, RYR2: ryanodine receptor isoform 2, SIRT3: sirtuin 3, MICOS: mt-cristae organizing system, ETC: electron transport chain, CPT1B: carnitine-O-palmitoyltransferase 1 muscle isoform, CPT2: carnitine-O-palmitoyltransferase isoform 2, AKAP: A-kinase anchoring protein, PP2A: calcineurin, DRP-1: dynamin-related protein 1, PLN: phospholamban, SERCA2a: sarco/endoplasmic reticulum calcium ATPase isoform 2a, TNNI3: troponin I, cardiac muscle, RI and RII: regulatory unit I and II, LTCC: L-type calcium channel, NCX-1: sodium/calcium exchanger 1, GPCR: G protein-coupled receptor, PLC: phospholipase C, DAG: diacylglycerol, MAPK: mitogen-activated protein kinase, JNK: c-Jun N-terminal kinase, Bcl-2: B-cell lymphoma 2, Bax: Bcl-2 associated X, BNIP3: Bcl-2 nineteen kilodalton interacting protein 3, AMPK: adenosine monophosphate-activated protein kinase, PGC-1α: peroxisome proliferator-activated receptor gamma coactivator-1 alpha. PDE4: phosphodiesterase 4, cAMP: cyclic adenosine monophosphate, and ADCY5: adenylate cyclase type 5.
Under normal conditions, mitochondrial ATP is generated primarily through the oxidation of fatty acid (60–90%) and glucose (10–40%). These substrates are processed in the cytoplasm and then are transported into the mitochondria as metabolic intermediates where they enter the tricarboxylic acid (TCA) cycle to produce reduced nicotinamide adenine dinucleotide (NADH), and flavin adenine dinucleotide (FADH2). NADH and FADH2 are then oxidized to NAD and FAD by the electron transport chain (ETC) system to generate a proton gradient across the IMM. The proton gradient is the driving force for ATP synthesis that takes place when protons flow via the ATP synthase (ETC complex V) into the mt-matrix. In the process, mt-reactive oxygen species (ROS) are physiologically generated, mainly from ETC complexes I and III in the form of oxygen radicals, which are reduced to hydrogen peroxide by the mitochondrial manganese superoxide dismutase. Hydrogen peroxide is then reduced to water by the mitochondrial or cytosolic antioxidant systems such as catalase, thioredoxin-peroxidase and glutathione-peroxidase. Calcium is a key second messenger that orchestrates the interplay of mt-redox and energetics with the excitation contraction coupling [19]. Increases in cytosolic and mt-calcium transients have been observed upon acute stimulation of the cardiomyocyte with isoproterenol and increases in energetic demand. This serves as a stimulus to activate enzymes in the TCA cycle and ETC complexes, such as pyruvate dehydrogenase, NAD+-dependent isocitrate dehydrogenase, α-ketoglutarate dehydrogenase and ETC complexes I and III [20], respectively. Therefore, the same signal that stimulates muscle contraction also stimulates ATP production to meet the higher energetic needs/demands, but at the expense of increased ROS generation. Crosstalk exists between calcium and ROS signaling systems. Together, they can regulate and fine tune cellular signaling networks [21]. Derangements in either of the aforementioned signaling systems would adversely affect the other and will have detrimental consequences on cellular fate and survival [21].
3. Calcium Signaling and Mitochondrial Oxidative Capacity
Calcium is an important second messenger involved in multiple cellular processes. It is crucial for the excitation-contraction coupling due to the calcium induced calcium release (CICR) phenomenon from the SR via the ryanodine receptors isoform 2 (RYR2) [22]. This phenomenon happens upon stimulation of the cardiomyocyte and opening of the L-type calcium channels (LTCC) leading to entry of small amount of calcium that triggers a large amount of calcium to be released from the junctional SR via RYR2 [23][24]. Then, calcium is pumped back into the SR via the enzyme sarco/endoplasmic reticulum ATPase (SERCA2a), promoting the initiation of the relaxation phase of the cardiac cycle, Figure 2. SERCA2a activity is regulated by phospholamban (PLN), which inhibits its activity [25][26]. The SR/ER and the mitochondria are important component of cardiac myocyte calcium cycling. They crosstalk with each other via calcium signaling through structural and functional interorganellar coupling [27]. The role of calcium in the ER is to control the calreticulin/calnexin cycle, which is required for proper protein folding, and cellular growth and development [28]. In mitochondria, calcium acts as a second messenger required to regulate enzymes of the TCA cycle and ETC complexes I and III. Calcium uptake into the mitochondria is orchestrated via the mostly abundant voltage dependent anion channel isoform 1 (VDAC1), compared to isoforms 2 and 3, at the OMM and is tightly regulated by the mitochondrial calcium uniporter (MCU) at the IMM [29][30], Figure 2. The MCU is a nodal regulator of calcium uptake into the mt-matrix and thus regulates mt-energetics and OXPHOS under cardiac stress conditions of high energetic demands [31]. Under basal conditions it seems that calcium can still enter the mt-matrix via MCU independent pathways to maintain cellular energetics, such as the ryanodine receptor isoform 1 (RYR1), however, this has not been well studied, Figure 2. VDAC1 is also an important player in mt-energetics/metabolism via its regulation of metabolites exchange across the mt-membranes and the ETC [32][33]. mt-calcium efflux is mediated via the sodium/calcium/lithium exchanger (NCLX) at the IMM into the IMS and then through VDAC1 into the cytoplasm [34]. Moreover, recent investigation has eluded to the importance of the hydrogen/calcium exchanger at the IMM, also known as the leucine zipper EF-hand containing transmembrane protein 1 (LETM1), to play an important role in regulating mt-calcium flux and energetics independent of MCU and NCLX [35]. Thus, the ER-mt crosstalk constitutes the functional unit that links calcium cycling to cardiac energetics. This ER-mt crosstalk relationship is tightly coordinated to maintain proper cellular function and is regulated by signal transduction pathways and B-cell lymphoma-2 (Bcl-2) family proteins at the ER-mt interface, Figure 2.
The ER-mt crosstalk relationship is perturbed in HF due to an alteration in cardiomyocyte ultrastructure and an imbalance in signaling and Bcl-2 family proteins regulation at the ER-mt interface. These changes take place at different stages of HF. Abnormal clustering of fragmented mitochondria is seen in pathological hypertrophy [36]. Upon transitioning to a decompensated state and SHF development, clustered fragmented mitochondria undergoing different stages of vacuolar degeneration are often seen and constitute the ultrastructural signature of HF [37][38], Figure 3. T-tubule dilatation and abnormal T-tubule-mt relationship [38] may alter calcium release from the dyadic cleft and the ER-mt crosstalk [39], Figure 3. The MAPK, JNK, which is activated in early pathological hypertrophy [36], plays detrimental role by promoting ER-mt calcium homeostasis dysregulation and apoptosis through the phosphorylation of Bcl-2 family proteins [36] and mitofusin 2(MFN2) [40]. Phosphorylation of PLN at Ser16 and dynamin related protein 1 (DRP1) at Ser637 were attenuated in a rat model of pressure overload induced moderate remodeling and early systolic dysfunction [9][41]. This leads to decreased calcium cycling and enhanced mt-fission and fragmentation, respectively, at the initial stages of cardiac remodeling and systolic dysfunction. It is not yet clear to whether these changes in PLN and DRP1 phosphorylation are related solely to an enhanced calcineurin activity [42] or also to an early decrease in PKA activity at the ER-mt interface subcellular compartment [43]. Eventually, the down-regulation of SERCA2a constitutes the signature for the progression to SHF and impaired calcium cycling [44][45] leading to depletion in ER calcium content and increase in cytosolic calcium, in addition to ER calcium leak via the RYR2 [46], Figure 4. Mitochondrial-matrix calcium overload and increased mt-ROS production are now being realized as important pathophysiological processes of mt-dysfunction and decreased oxidative capacity in HF [34][47][48]. It has been shown that MCU expression level and mt-calcium uptake were attenuated in diabetic cardiomyopathy [49]. This paradox in decreased MCU expression level and mt-matrix calcium overload could be explained by activation of signaling pathways that can modulate mt-calcium uptake and/or efflux proteins at the OMM and IMM via a post-translational modification (PTM) mechanism, Figure 4. This area may need to be elucidated in future studies. Moreover, mt-calcium efflux kinetics is a much slower process than mt-calcium uptake [50][51], owing to the possible explanation of increases in mt-matrix calcium during acute, such as isoproterenol stimulation, and chronic cardiac stress conditions, as is in HF. In the former, increases in mt-matrix calcium acts as a stimulant for ATP production; in the latter, mt-matrix calcium overload is detrimental leading to mt-vacuolar degeneration and increased ROS and apoptosis. These differences between acute vs. chronic cardiac stress are likely related to distinct signaling pathways and molecular mechanisms modulating mt-calcium homeostasis and mt-function that are not entirely known. Future work is needed in this area to elucidate molecular mechanisms regulating mt-calcium and metabolism in acute versus chronic cardiac stress.
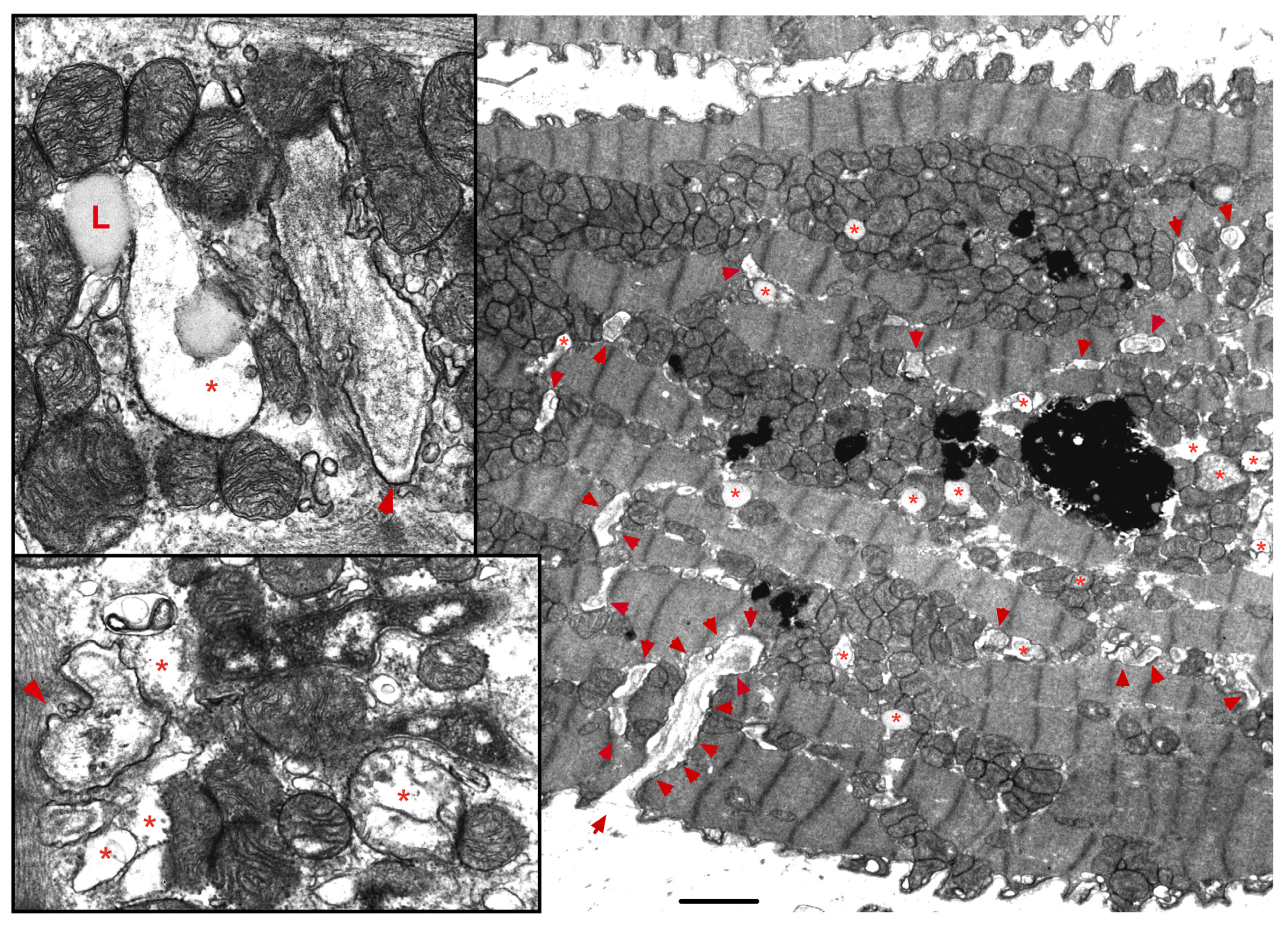
Figure 3. Ultrastructure of the failing myocardium. Transmission electron photomicrograph of myocardium from human failing heart showing typical clustering and fragmentation of intermyofibrillar mitochondria. Red asterisk showing mitochondria at advanced stage of vacuolar degeneration. There is perturbed relationship between the intermyofibrillar mitochondria and the surrounding sarcomeres and T-tubules (red arrowhead). Note that the T-tubules are dilated and that they are pushed to the edge of the clustered mitochondria, instead of being interspersed between adjacent mitochondria as seen in a normal myocardium. Moreover, numerous lysosomes (black, dense lamellar structures) are found interspersed between clustered mitochondria. Image 5k × magnified, scale bar 2 μm. Zoomed images showing in more detail the changes in mitochondrial cristae morphology and decrease in their density that occur in heart failure. Abbreviation: L: lipid.
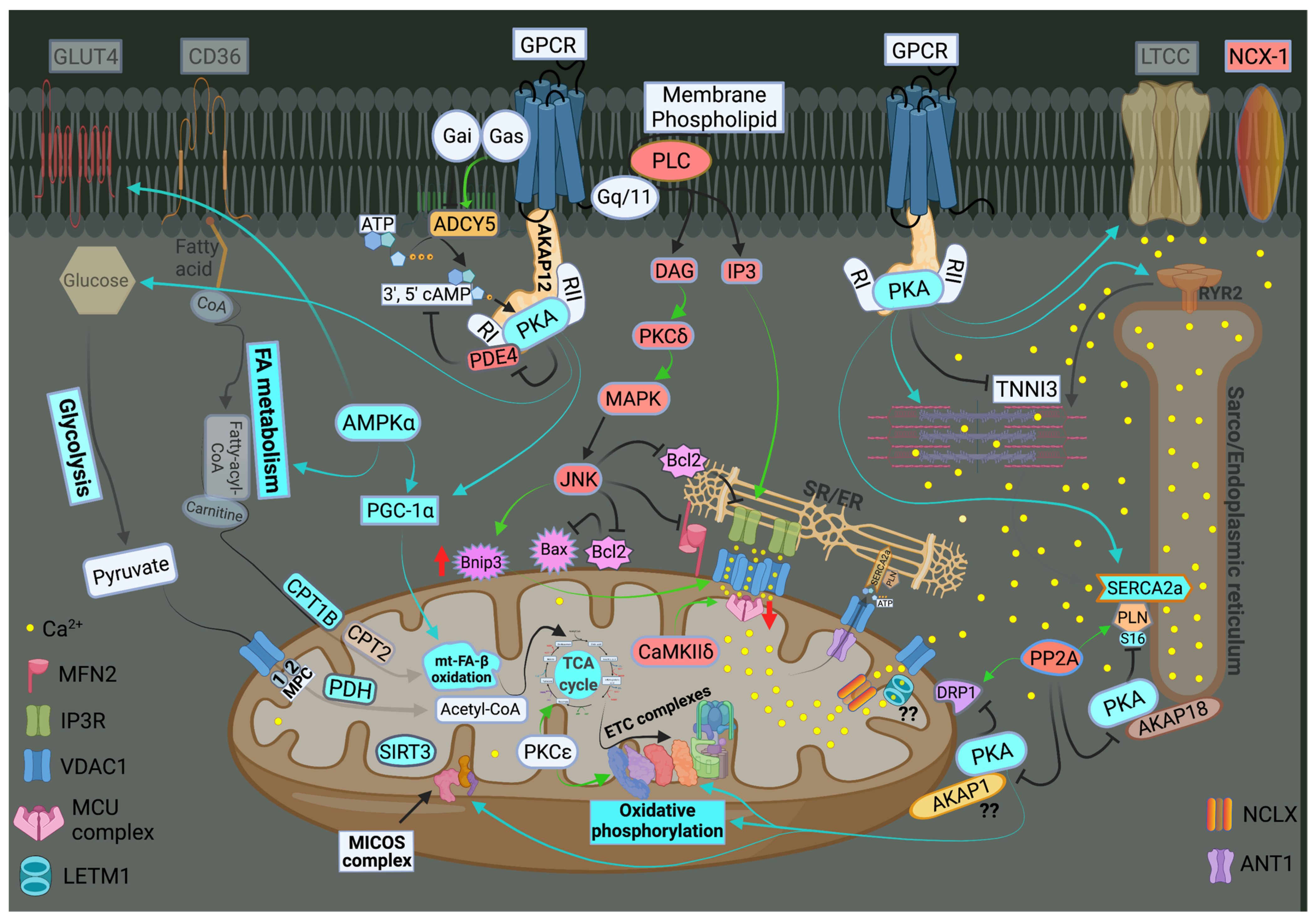
Figure 4. Schematic drawing showing metabolic remodeling, calcium cycling dysregulation and altered signaling in heart failure. Green arrows: promote signaling or activity. Turquoise arrows, rectangle or circle denotes decrease in signaling, activity or expression. Red rectangle or circle denotes increase in activity or expression. The deeper the intensity of the turquoise or red color, the higher is the degree in downregulation or upregulation, respectively, of protein activity or expression. Red up and down arrows indicate an increase in BNIP3 expression and a decrease in MCU expression, respectively. FA metabolism and mt-FA-β-oxidation, glycolysis, TCA and OXPHOS are attenuated in HF. SIRT3 expression is decreased in HF leading to mt-protein hyperacetylation. Enhanced MAPK (JNK) signaling inhibits Bcl-2 and MFN2 leading to ER-mt calcium dysregulation, mt-fission and apoptosis. Perturbations in calcium cycling and ER-mt tethering are evident in HF. ER calcium is depleted due to an enhanced ER calcium leak via RYR2, and a decrease in ER calcium uptake via SERCA2a leading to an increase in cytosolic calcium. Overall PKA signaling is attenuated in HF. Most importantly, decrease in PKA signaling and enhanced calcineurin activity at the ER-mt interface leads to (1) enhanced mt-fission, through recruitment of DRP1 to the mt-outer membrane; (2) decrease in ETC activity and OXPHOS; and (3) decreased in p-S16-PLN leading to further impairment in SERCA2a activity. Increases in IP3 abundance and BNIP3 expression lead to enhanced calcium release via the IP3R and enhanced calcium uptake via the VDAC1 channels, respectively. This is due to conformational change (oligomerization) of VDAC1. Enhanced CaMKIIδ expression or activity enhances mt-calcium uptake via MCU complex leading to mt-matrix calcium overload and mt-dysfunction. Mitochondrial calcium efflux is likely to be affected as well. It is unclear whether there is post-translational modification (.) and decrease in activity or expression of LETM1 and AKAP1 in HF. Abbreviations are listed under Figure 2.
Previous work showed that recombinant expression of VDAC1 enhanced ER-mt contact sites and calcium transfer into the mitochondria and apoptosis [52][53]. In HF, VDAC1 expression does not change compared with normal heart. However, VDAC1 oligomerization or modulation by the Bcl-2 family proteins [48], as discussed below, or in response to apoptotic stimuli [54], may alter calcium entry into the IMS and subsequently into the mt-matrix leading to mt-matrix calcium overload, mt-dysfunction and apoptosis, Figure 4. The exact molecular mechanism that promotes VDAC1 oligomerization in HF is not known. Moreover, previous work has shown that calcium uptake via MCU is enhanced by its PTM by the mt-CaMKII [55][56], Figure 4. Conditional NCLX knockout in mouse heart has been shown to be lethal, with less than 13% of affected animals surviving beyond day 14 [57]. These findings point to the importance of the NCLX in mt-calcium efflux. Recently, it has been shown that MCU over-expression rescues inotropy and reverses HF by reducing SR calcium leak [58]. Overall, these findings hint to the complexity of ER-mt calcium homeostasis regulation and function. Whether other modes of mt-calcium entry or other mechanisms regulating mt-calcium uptake and efflux at the IMM exist, need to be elucidated in future studies.
Previous work has shown that mt-biogenesis and oxidative capacity were preserved in patients with HF and preserved ejection fraction (HFpEF) [38] or enhanced in pathological compensated hypertrophy [9][59]. A decline in mt-content and oxidative capacity has been observed in rodent models upon transitioning from a compensated state to moderate remodeling and early systolic dysfunction [9]. At the molecular level, these findings are mirrored by a decrease in PGC-1α expression upon transitioning from compensated hypertrophy to early systolic dysfunction [9]. Cytochrome c oxidase activity is attenuated in early systolic dysfunction along with a decrease in expression of the ETC complexes I and IV [41]. Transitioning to overt SHF and HFrEF is marked by a further decrease in mt-biogenesis and PGC-1α expression and signaling, along with a decrease in expression of the ETC complexes, Figure 4. These findings were observed in an animal model of SHF and in human HFrEF [38][59]. Another mechanism of decreased oxidative capacity in HF is phosphorylation (discussed below) and hyperacetylation of mt-proteins. Increased acetylation of mt-proteins involved in mt-FA-β-oxidation, TCA cycle and ETC complexes has been observed in the early stages of HF in a pressure overload mouse model of transverse aortic constriction (TAC) and in end-stage human HFrEF [60][61]. Hyperacetylation of mt-proteins is speculated to be related to the presence of excessive acyl-CoAs and reduced protein deacetylation by the sirtuin family of NAD+-dependent deacetylases, mainly SIRT3 [62], and SIRT5. SIRT3 is the predominant mt-deacetylase isoform in the heart. SIRT3-/- mice develop contractile dysfunction at 24 weeks of age [63]. Myocardial SIRT3 expression was increased in cardiac hypertrophy following TAC in mice [64] and then was downregulated at HF development [65][66]. Similarly, SIRT3 expression was decreased in the human failing heart [67]. PGC-1α was found to regulate SIRT3 expression through the AMPK-PGC-1α-SIRT3 axis in skeletal muscle [68]. Moreover, SIRT3 downregulation seems to coincide with the decrease in PGC-1α expression in rodent and human HF [69]. Thus, a decline in mt-oxidative capacity in early systolic dysfunction is mainly related to an increase in mt-ROS and a decrease in activity of the OXPHOS machinery. Transitioning to overt SHF is accompanied by decreases in mt-content and mt-biogenesis, including expression of the ETC complexes, as well as by an increase in mt-ROS and PTM, therefore, phosphorylation and hyperacetylation, of the OXPHOS machinery.
References
- Neubauer, S. The failing heart—An engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151.
- D’Erchia, A.M.; Atlante, A.; Gadaleta, G.; Pavesi, G.; Chiara, M.; De Virgilio, C.; Manzari, C.; Mastropasqua, F.; Prazzoli, G.M.; Picardi, E.; et al. Tissue-specific mtDNA abundance from exome data and its correlation with mitochondrial transcription, mass and respiratory activity. Mitochondrion 2015, 20, 13–21.
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726.
- Galluzzi, L.; Kepp, O.; Trojel-Hansen, C.; Kroemer, G. Mitochondrial Control of Cellular Life, Stress, and Death. Circ. Res. 2012, 111, 1198–1207.
- Sack, M.N.; Rader, T.A.; Park, S.; Bastin, J.; McCune, S.A.; Kelly, D.P. Fatty acid oxidation enzyme gene expression is down-regulated in the failing heart. Circulation 1996, 94, 2837–2842.
- Doenst, T.; Pytel, G.; Schrepper, A.; Amorim, P.; Färber, G.; Shingu, Y.; Mohr, F.W.; Schwarzer, M. Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc. Res. 2009, 86, 461–470.
- Stride, N.; Larsen, S.; Hey-Mogensen, M.; Sander, K.; Lund, J.T.; Gustafsson, F.; Køber, L.; Dela, F. Decreased mitochondrial oxidative phosphorylation capacity in the human heart with left ventricular systolic dysfunction. Eur. J. Heart Fail. 2013, 15, 150–157.
- Barger, P.M.; Brandt, J.M.; Leone, T.C.; Weinheimer, C.J.; Kelly, D.P. Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J. Clin. Investig. 2000, 105, 1723–1730.
- Chaanine, A.H.; Sreekumaran Nair, K.; Bergen, R.H.; Klaus, K.; Guenzel, A.J.; Hajjar, R.J.; Redfield, M.M. Mitochondrial Integrity and Function in the Progression of Early Pressure Overload-Induced Left Ventricular Remodeling. J. Am. Heart Assoc. 2017, 6, e005869.
- Huss, J.M.; Kelly, D.P. Nuclear Receptor Signaling and Cardiac Energetics. Circ. Res. 2004, 95, 568–578.
- Sutanto, H.; Lyon, A.; Lumens, J.; Schotten, U.; Dobrev, D.; Heijman, J. Cardiomyocyte calcium handling in health and disease: Insights from in vitro and in silico studies. Prog. Biophys. Mol. Biol. 2020, 157, 54–75.
- Kurland, C.G.; Andersson, S.G. Origin and evolution of the mitochondrial proteome. Microbiol. Mol. Biol. Rev. MMBR 2000, 64, 786–820.
- D’Souza, A.R.; Minczuk, M. Mitochondrial transcription and translation: Overview. Essays Biochem. 2018, 62, 309–320.
- Taanman, J.-W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta BBA Bioenerg. 1999, 1410, 103–123.
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714.
- Hoppel, C.L.; Tandler, B.; Fujioka, H.; Riva, A. Dynamic organization of mitochondria in human heart and in myocardial disease. Int. J. Biochem. Cell Biol. 2009, 41, 1949–1956.
- Kuznetsov, A.V.; Margreiter, R. Heterogeneity of Mitochondria and Mitochondrial Function within Cells as Another Level of Mitochondrial Complexity. Int. J. Mol. Sci. 2009, 10, 1911–1929.
- Boncompagni, S.; Rossi, A.E.; Micaroni, M.; Beznoussenko, G.V.; Polishchuk, R.; Dirksen, R.T.; Protasi, F. Mitochondria Are Linked to Calcium Stores in Striated Muscle by Developmentally Regulated Tethering Structures. Mol. Biol. Cell 2009, 20, 1058–1067.
- Bertero, E.; Maack, C. Metabolic remodelling in heart failure. Nat. Rev. Cardiol. 2018, 15, 457–470.
- McCormack, J.G.; Halestrap, A.P.; Denton, R.M. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 1990, 70, 391–425.
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271.
- Fabiato, A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 1983, 245, C1–C14.
- Williams, G.S.; Huertas, M.A.; Sobie, E.A.; Jafri, M.S.; Smith, G.D. A Probability Density Approach to Modeling Local Control of Calcium-Induced Calcium Release in Cardiac Myocytes. Biophys. J. 2007, 92, 2311–2328.
- Williams, G.S.B.; Huertas, M.A.; Sobie, E.A.; Jafri, M.S.; Smith, G.D. Moment Closure for Local Control Models of Calcium-Induced Calcium Release in Cardiac Myocytes. Biophys. J. 2008, 95, 1689–1703.
- Minamisawa, S.; Hoshijima, M.; Chu, G.; Ward, C.A.; Frank, K.; Gu, Y.; Martone, M.E.; Wang, Y.; Ross, J.; Kranias, E.G.; et al. Chronic Phospholamban–Sarcoplasmic Reticulum Calcium ATPase Interaction Is the Critical Calcium Cycling Defect in Dilated Cardiomyopathy. Cell 1999, 99, 313–322.
- Gustavsson, M.; Verardi, R.; Mullen, D.G.; Mote, K.R.; Traaseth, N.J.; Gopinath, T.; Veglia, G. Allosteric regulation of SERCA by phosphorylation-mediated conformational shift of phospholamban. Proc. Natl. Acad. Sci. USA 2013, 110, 17338–17343.
- Rimessi, A.; Pedriali, G.; Vezzani, B.; Tarocco, A.; Marchi, S.; Wieckowski, M.; Giorgi, C.; Pinton, P. Interorganellar calcium signaling in the regulation of cell metabolism: A cancer perspective. Semin. Cell Dev. Biol. 2019, 98, 167–180.
- Michalak, M.; Parker, J.R.; Opas, M. Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum. Cell Calcium 2002, 32, 269–278.
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472.
- Wu, Y.; Rasmussen, T.P.; Koval, O.M.; Joiner, M.L.; Hall, D.D.; Chen, B.; Luczak, E.D.; Wang, Q.; Rokita, A.G.; Wehrens, X.H.; et al. The mitochondrial uniporter controls fight or flight heart rate increases. Nat. Commun. 2015, 6, 6081.
- Luongo, T.; Lambert, J.; Yuan, A.; Zhang, X.; Gross, P.; Song, J.; Shanmughapriya, S.; Gao, E.; Jain, M.; Houser, S.R.; et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015, 12, 23–34.
- Abu-Hamad, S.; Sivan, S.; Shoshan-Barmatz, V. The expression level of the voltage-dependent anion channel controls life and death of the cell. Proc. Natl. Acad. Sci. USA 2006, 103, 5787–5792.
- Vander Heiden, M.G.; Chandel, N.S.; Li, X.X.; Schumacker, P.T.; Colombini, M.; Thompson, C.B. Outer mitochondrial mem-brane permeability can regulate coupled respiration and cell survival. Proc. Natl. Acad. Sci. USA 2000, 97, 4666–4671.
- Finkel, T.; Menazza, S.; Holmstrom, K.M.; Parks, R.J.; Liu, J.; Sun, J.; Liu, J.; Pan, X.; Murphy, E. The ins and outs of mitochondrial calcium. Circ. Res. 2015, 116, 1810–1819.
- Doonan, P.J.; Chandramoorthy, H.C.; Hoffman, N.E.; Zhang, X.; Cárdenas, C.; Shanmughapriya, S.; Rajan, S.; Vallem, S.; Chen, X.; Foskett, J.K.; et al. LETM1-dependent mitochondrial Ca2+ flux modulates cellular bioenergetics and proliferation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 4936–4949.
- Chaanine, A.H.; Jeong, D.; Liang, L.; Chemaly, E.R.; Fish, K.; Gordon, R.E.; Hajjar, R.J. JNK modulates FOXO3a for the ex-pression of the mitochondrial death and mitophagy marker BNIP3 in pathological hypertrophy and in heart failure. Cell Death Dis. 2012, 3, 265.
- Chaanine, A.H. Morphological Stages of Mitochondrial Vacuolar Degeneration in Phenylephrine-Stressed Cardiac Myocytes and in Animal Models and Human Heart Failure. Medicina 2019, 55, 239.
- Chaanine, A.H.; Joyce, L.D.; Stulak, J.M.; Maltais, S.; Joyce, D.L.; Dearani, J.A.; Klaus, K.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. Mitochondrial Morphology, Dynamics, and Function in Human Pressure Overload or Ischemic Heart Disease with Preserved or Reduced Ejection Fraction. Circ. Heart Fail. 2019, 12, e005131.
- Louch, W.E.; Sejersted, O.M.; Swift, F. There goes the neighborhood: Pathological alterations in T-tubule morphology and consequences for cardiomyocyte Ca2+ handling. J. Biomed. Biotechnol. 2010, 2010, 503906.
- Leboucher, G.P.; Tsai, Y.C.; Yang, M.; Shaw, K.C.; Zhou, M.; Veenstra, T.D.; Glickman, M.H.; Weissman, A.M. Stress-Induced Phosphorylation and Proteasomal Degradation of Mitofusin 2 Facilitates Mitochondrial Fragmentation and Apoptosis. Mol. Cell 2012, 47, 547–557.
- Chaanine, A.H.; Kohlbrenner, E.; Gamb, S.I.; Guenzel, A.J.; Klaus, K.; Fayyaz, A.U.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am. J. Physiol. Circ. Physiol. 2016, 311, H1540–H1559.
- Cereghetti, G.M.; Stangherlin, A.; Martins de Brito, O.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphory-lation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808.
- Chang, C.-R.; Blackstone, C. Cyclic AMP-dependent Protein Kinase Phosphorylation of Drp1 Regulates Its GTPase Activity and Mitochondrial Morphology. J. Biol. Chem. 2007, 282, 21583–21587.
- Bers, D.M.; Despa, S.; Bossuyt, J. Regulation of Ca2+ and Na+ in Normal and Failing Cardiac Myocytes. Ann. N. Y. Acad. Sci. 2006, 1080, 165–177.
- Hajjar, R.J. Potential of gene therapy as a treatment for heart failure. J. Clin. Investig. 2013, 123, 53–61.
- Marks, A.R. Calcium cycling proteins and heart failure: Mechanisms and therapeutics. J. Clin. Investig. 2013, 123, 46–52.
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394.
- Chaanine, A.H.; Gordon, R.E.; Kohlbrenner, E.; Benard, L.; Jeong, D.; Hajjar, R.J. Potential role of BNIP3 in cardiac remodeling, myocardial stiffness, and endoplasmic reticulum: Mitochondrial calcium homeostasis in diastolic and systolic heart failure. Circ. Heart Fail. 2013, 6, 572–583.
- Suarez, J.; Cividini, F.; Scott, B.; Lehmann, K.; Diaz-Juarez, J.; Diemer, T.; Dai, A.; Jain, M.; Dillmann, W.H. Restoring mitochondrial calcium uniporter expression in diabetic mouse heart improves mitochondrial calcium handling and cardiac function. J. Biol. Chem. 2018, 293, 8182–8195.
- Lu, X.; Ginsburg, K.S.; Kettlewell, S.; Bossuyt, J.; Smith, G.L.; Bers, D.M. Measuring Local Gradients of Intramitochondrial in Cardiac Myocytes During Sarcoplasmic Reticulum Ca2+ Release. Circ. Res. 2013, 112, 424–431.
- Maack, C.; Cortassa, S.; Aon, M.A.; Ganesan, A.N.; Liu, T.; O’Rourke, B. Elevated Cytosolic Na+ Decreases Mitochondrial Ca2+ Uptake During Excitation-Contraction Coupling and Impairs Energetic Adaptation in Cardiac Myocytes. Circ. Res. 2006, 99, 172–182.
- Rapizzi, E.; Pinton, P.; Szabadkai, G.; Wieckowski, M.; Vandecasteele, G.; Baird, G.; Tuft, R.A.; Fogarty, K.E.; Rizzuto, R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 2002, 159, 613–624.
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285.
- Keinan, N.; Tyomkin, D.; Shoshan-Barmatz, V. Oligomerization of the Mitochondrial Protein Voltage-Dependent Anion Channel Is Coupled to the Induction of Apoptosis. Mol. Cell. Biol. 2010, 30, 5698–5709.
- Joiner, M.-L.A.; Koval, O.; Li, J.; He, B.J.; Allamargot, C.; Gao, Z.; Luczak, E.D.; Hall, D.; Fink, B.D.; Chen, B.; et al. CaMKII determines mitochondrial stress responses in heart. Nature 2012, 491, 269–273.
- Nguyen, E.K.; Koval, O.M.; Noble, P.; Broadhurst, K.; Allamargot, C.; Wu, M.; Strack, S.; Thiel, W.H.; Grumbach, I.M. CaMKII (Ca(2+)/Calmodulin-Dependent Kinase II) in Mitochondria of Smooth Muscle Cells Controls Mitochondrial Mobility, Migration, and Neointima Formation. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1333–1345.
- Luongo, T.; Lambert, J.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; van Berlo, J.; et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 2017, 545, 93–97.
- Liu, T.; Yang, N.; Sidor, A.; O’Rourke, B. MCU Overexpression Rescues Inotropy and Reverses Heart Failure by Reducing SR Ca2+ Leak. Circ. Res. 2021, 128, 1191–1204.
- Rosca, M.G.; Tandler, B.; Hoppel, C.L. Mitochondria in cardiac hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2012, 55, 31–41.
- Horton, J.L.; Martin, O.J.; Lai, L.; Riley, N.M.; Richards, A.L.; Vega, R.B.; Leone, T.C.; Pagliarini, D.J.; Muoio, D.M.; Bedi, K.C.; et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight 2016, 2, e84897.
- Lee, C.F.; Chavez, J.D.; Garcia-Menendez, L.; Choi, Y.S.; Roe, N.D.; Chiao, Y.A.; Edgar, J.S.; Goo, Y.A.; Goodlett, D.R.; Bruce, J.E.; et al. Normalization of NAD + Redox Balance as a Therapy for Heart Failure. Circulation 2016, 134, 883–894.
- Chen, T.; Liu, J.; Li, N.; Wang, S.; Liu, H.; Li, J.; Zhang, Y.; Bu, P. Correction: Mouse SIRT3 Attenuates Hypertrophy-Related Lipid Accumulation in the Heart through the Deacetylation of LCAD. PLoS ONE 2016, 11, e0155173.
- Koentges, C.; Pfeil, K.; Schnick, T.; Wiese, S.; Dahlbock, R.; Cimolai, M.C.; Meyer-Steenbuck, M.; Cenkerova, K.; Hoffmann, M.M.; Jaeger, C.; et al. SIRT3 deficiency impairs mitochondrial and contractile function in the heart. Basic Res. Cardiol. 2015, 110, 36.
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771.
- Pillai, V.B.; Bindu, S.; Sharp, W.W.; Fang, Y.H.; Kim, G.; Gupta, M.; Samant, S.; Gupta, M.P. Sirt3 protects mitochondrial DNA damage and blocks the development of doxorubicin-induced cardiomyopathy in mice. Am. J. Physiol. Circ. Physiol. 2016, 310, H962–H972.
- Pillai, V.B.; Kanwal, A.; Fang, Y.H.; Sharp, W.W.; Samant, S.; Arbiser, J.; Gupta, M.P. Honokiol, an activator of Sirtuin-3 (SIRT3) preserves mitochondria and protects the heart from doxorubicin-induced cardiomyopathy in mice. Oncotarget 2017, 8, 34082–34098.
- Koentges, C.; Bode, C.; Bugger, H. SIRT3 in Cardiac Physiology and Disease. Front. Cardiovasc. Med. 2016, 3, 38.
- Shi, L.; Zhang, T.; Zhou, Y.; Zeng, X.; Ran, L.; Zhang, Q.; Zhu, J.; Mi, M. Dihydromyricetin improves skeletal muscle insulin sensitivity by inducing autophagy via the AMPK-PGC-1α-Sirt3 signaling pathway. Endocrine 2015, 50, 378–389.
- Oka, S.-I.; Sabry, A.D.; Cawley, K.M.; Warren, J.S. Multiple Levels of PGC-1α Dysregulation in Heart Failure. Front. Cardiovasc. Med. 2020, 7, 2.
More
Information
Subjects:
Cell Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
20 Oct 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No