Castration-resistant prostate cancer (CRPC) is a clinical challenge in treatment because of its aggressive nature and resistance to androgen deprivation therapy. Topoisomerase II catalytic inhibitors have been suggested as a strategy to overcome these issues. We previously reported AK-I-190 as a novel topoisomerase II inhibitor. In this study, the mechanism of AK-I-190 was clarified using various types of spectroscopic and biological evaluations. AK-I-190 showed potent topoisomerase II inhibitory activity through intercalating into DNA without stabilizing the DNA-enzyme cleavage complex, resulting in significantly less DNA toxicity than etoposide, a clinically used topoisomerase II poison. AK-I-190 induced G1 arrest and effectively inhibited cell proliferation and colony formation in combination with paclitaxel in an androgen receptor–negative CRPC cell line.
1. Introduction
Prostate cancer (PCa) is one of the most common types of cancer in elderly men
[1]. This type of cancer is driven by the androgen receptor (AR) signaling pathway, and therefore AR-targeting drugs in androgen-deprivation therapy are currently the primary therapeutic option for PCa
[2]. AR-targeting treatment results in significant reduction of tumoral growth and progression at all stages of PCa
[3]. However, in a few cases, the tumor adapts to AR suppression through persistent activation of AR signaling without ligand or complete elimination in AR expression, leading to recurrent and metastatic cancer
[4][5].
After exposure to androgen-deprivation therapy, AR-positive PCa cells undergo lineage plasticity and epigenetic reprograming
[6]. During this process, the development of stem cell likeness transforms PCa cells to gain androgen independence in cellular growth, so-called castration resistance
[7]. Castration-resistant prostate cancer (CRPC) has emerged as a major obstacle in PCa treatment. It is strongly believed that alternative oncogenic signaling pathways are activated to bypass androgen dependency, which leads to CRPC development. Therefore, to overcome castration resistance and effectively manage PCa, a therapeutic strategy involving additional targets in addition to targeting AR signaling is required.
Topoisomerase II is an enzyme that resolves topological issues in the process of replication, transcription, recombination, chromosomal condensation and segregation
[8]. Because of the highly proliferative nature of cancer cells, topoisomerase II inhibitors, such as etoposide, doxorubicin and mitoxantrone, have long been used in diverse types of solid tumors. These topoisomerase II inhibitors have also been suggested for PCa treatment. Mitoxantrone was approved by the FDA in 1996 for the treatment of PCa, and several studies have shown clinical benefits of etoposide in combination regimens for the treatment of patients with CRPC
[9][10][11].
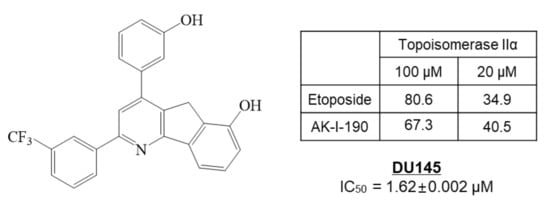
In our previous study, we synthesized 172 new halogenated 2,4-diphenyl Indeno[1,2-
b]pyridinol derivatives as topoisomerase inhibitors
[12]. Among the inhibitors, AK-I-190, 2-(3-trifluorophenyl)-4-(3-hydroxyphenyl)-5
H-indeno[1,2-
b]pyridin-6-ol, showed potent topoisomerase II inhibition with anti-proliferative activity in the DU145 AR-negative PCa cell line (
Figure 1). In this study, we clarified the molecular mechanism of AK-I-190 as a DNA intercalating catalytic inhibitor of topoisomerase II. We further evaluated the anti-proliferative and pro-apoptotic activities of AK-I-190 in AR-independently growing PCa cells. Our results suggest the clinical relevance and efficacy of the topoisomerase II catalytic inhibitor in AR-negative PCa.
Figure 1. The structure and topoisomerase II inhibitory and anti-proliferative activities of AK-I-190 (2-(3-trifluorophenyl)-4-(3-hydroxyphenyl)-5
H-indeno[1,2-
b]pyridin-6-ol) in DU145 prostate cancer cells.
2. Topoisomerase II Inhibitory Activity of AK-I-190 as a Catalytic Inhibitor
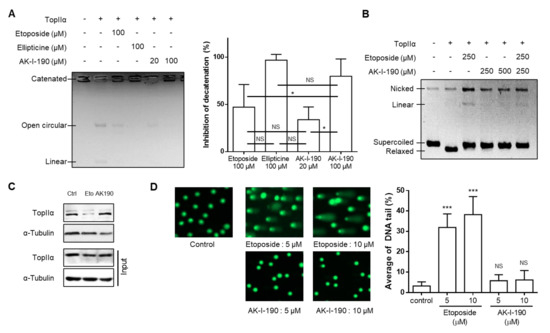
We evaluated the DNA topoisomerase II inhibitory activity of AK-I-190, synthesized as previously reported
[12], using the kinetoplast DNA (kDNA) decatenation assay. This experimental method is used for the assessment of topoisomerase II-specific decatenating ability
[13]. DNA topoisomerase II, but not DNA topoisomerase I, has the ability to decatenate DNA due to its ability to cleave DNA double strands simultaneously, which is essential for cells that hyper-proliferate through the process of mitosis and cytokinesis
[9]. Our results showed that AK-I-190 treatment retained kDNA in the catenated form (
Figure 2A). Notably, the same concentration of AK-I-190 (100 μM) showed better or equivalent inhibitory activity compared with the known topoisomerase II inhibitors etoposide and ellipticine. We next evaluated whether AK-I-190 was capable of stabilizing the DNA-enzyme complex. Stabilization of the topoisomerase-DNA cleavage complex produces linear DNA, which is a cause of adverse effects induced by topoisomerase poison, including etoposide
[14]. In the cleavage complex assay, the linear form of DNA was not generated by AK-I-190, whereas etoposide treatment resulted in a linear DNA band (
Figure 2B).
Figure 2. AK-I-190 inhibits topoisomerase II inhibition as a catalytic inhibitor. (A) kDNA decatenation assay of AK-I-190. The graph on the right shows quantification of the values measured in the gel. (B) Cleavage complex assay. Supercoiled DNA (pBR322) was pre-incubated with DNA topoisomerase IIα and the indicated compound was added. After incubation and agarose gel electrophoresis, bands of linear DNA were analyzed. (C) Band depletion assay. Formation of topoisomerase IIα–DNA cleavage complex was assessed using the band depletion assay. Cells treated with vehicle, etoposide, or AK-I-190 were lysed with denaturing buffer and centrifuged. DNA-unbound topoisomerase IIα was evaluated in the supernatant (upper panel). The whole cell lysate in the same experimental condition was used as input. (D) Alkaline comet assay. Comet slides were electrophoresed at high pH and stained with a SYBR gold solution. The comet tails were observed by fluorescence microscopy. Quantification of DNA tails was conducted using Komet 5.5 software. The intensities of comet tails versus comet heads reflected the extent of DNA breaks (n = 50). * p < 0.05 and *** p < 0.001 vs. control; NS, not significant.
As our results showed that AK-I-190 effectively inhibited enzymatic activity without induction of linearized DNA, a band depletion assay was conducted to determine whether AK-I-190 was a poison or a catalytic inhibitor. Treatment with etoposide, but not AK-I-190, resulted in a significant decrease in free topoisomerase IIα level, indicating that topoisomerase IIα covalently bound to DNA was increased in etoposide-treated cells (Figure 2C). Truncated DNA induced by stabilization of the enzyme-DNA complex was confirmed by alkaline comet assay (Figure 2D). Unlike etoposide, AK-I-190 did not result in an increase in DNA tail compared with controls. Collectively, these results indicate that AK-I-190 is a catalytic inhibitor of topoisomerase II rather than a poison.
3. DNA Intercalation of AK-I-190
For a deeper understanding of the mechanism of topoisomerase II inhibitory activity, we analyzed AK-I-190 using various spectroscopic methods. There are several known modes of action of topoisomerase II–targeting drugs, including stabilizing the cleavage complex, inhibiting ATPase activity of topoisomerase II, chelating metal ions essential for topoisomerase II function, and inhibiting DNA access to topoisomerase II by binding to DNA. As our results indicate that AK-I-190 is a catalytic inhibitor of topoisomerase II, we evaluated AK-I-190 activity in each mode of action except for cleavage complex stabilization. AK-I-190 did not inhibit topoisomerase II ATPase activity and had no chelating ability
[14] (
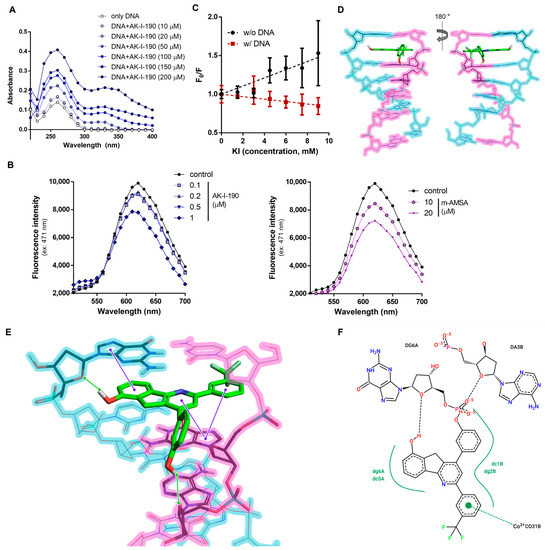
Supplementary Figure S1). For the evaluation of DNA binding, the absorption spectra of DNA with diverse concentrations of AK-I-190 were analyzed (
Figure 3A). Titrating AK-I-190 in DNA solution increased absorbance, confirming that AK-I-190 was bound to DNA. The absorbance values of 200 μM calf thymus DNA (ct-DNA) and 200 μM AK-I-190 at 260 nm wavelength were 0.174 and 0.075, respectively. The absorbance of a mixture of the same concentration of ct-DNA and AK-I-190 (200 μM each) increased to 0.350 at 260 nm. This result indicates that AK-I-190 interacts with DNA, thereby attenuating enzymatic activity of topoisomerase II.
Figure 3. DNA intercalation of AK-I-190. (
A) Interaction of AK-I-190 with ct-DNA was monitored using UV/vis spectroscopy. UV/vis spectrum for ct-DNA (200 μM) in the presence of various AK-I-190 concentrations (0–200 μM) in 10 mM Tris buffer (pH 7.4). (
B) Fluorescence spectra during titration of AK-I-190 to the complex of ct-DNA with EtBr (intercalator). The EtBr-DNA complex was excited at 471 nm and emission spectra were recorded from 500 nm to 700 nm. Fluorescence intensity decreased with subsequent addition of AK-I-190 (left) and m-AMSA (right). (
C) KI quenching experiment. Stern–Volmer plot of fluorescence quenching of AK-I-190 (100 μM) by increasing KI concentration (0–9 mM) in 10 mM Tris buffer (pH 7.4) in the presence (red) and absence (black) of ct-DNA. (
D,
E) Molecular docking study of AK-I-190. Docking studies of AK-I-190 were carried out using the X-ray crystal structure of human DNA complexed with ellipticine as a template (PDB code, 1Z3F) using the Flare program. AK-I-190 potentially binds to DNA double helix by intercalation between base pairs. Each DNA strand is shown as blue and pink highlighted lines. Residues participating in the interaction are indicated by bold sticks and AK-I-190 is shown by capped sticks. Both are colored according to atom (carbon, black or green; oxygen, red; nitrogen, blue; phosphorus, cyan; fluoride, light green). Expected hydrogen bonds are indicated as green solid lines and π–π interactions are represented as purple solid lines. (
F) Two-dimensional diagram of the interaction between AK-I-190 and the DNA double helical structure. The diagram was created using Pose View in
proteins.plus/ (accessed on 20 August 2021).
We next examined the binding mode of AK-I-190 to DNA using EtBr displacement and KI quenching assays. AK-I-190 was confirmed to interact with DNA in an intercalating manner similar to that of m-AMSA, a known DNA intercalating agent (
Figure 3B,C)
[15]. The fluorescence quenching ability of iodide ions on AK-I-190 was evaluated in the absence and presence of DNA co-treatment. The fluorescence of the DNA intercalating agent tends to weaken the fluorescence quenching ability of iodide ions under the DNA co-treatment condition because of the intercalating action, and thus the Fo/F ratio value tends to decrease as the KI concentration increases. The Stern–Volmer quenching constant (K
SV) value of iodide ion was 52.99 M
−1 in the absence of ct-DNA, whereas its fluorescence-quenching ability was completely diminished by ct-DNA addition. AK-I-190 was revealed to be a DNA intercalating agent and not a groove binder.
DNA intercalation of AK-I-190 was further analyzed using a docking study (
Figure 3D–F). The results revealed that two hydroxyl groups of AK-I-190 formed hydrogen bonds with each chain of DNA, and the ring structure participated in multiple π–π interactions with purine and pyrimidine rings of DNA. The flat structure of AK-I-190 was responsible for the DNA intercalation and the additional hydrogen bonds were for anchorage.
4. Discussion
In this study, we clarified molecular mechanism of AK-I-190 as a DNA intercalating catalytic inhibitor of topoisomerase II. We first assessed whether AK-I-190 is a poison or catalytic inhibitor of topoisomerase II. AK-I-190 did not generate the linear form of DNA in cleavage complex assay and sustained DNA-unbound topoisomerase II in band depletion assay. These results revealed that AK-I-190 is a catalytic inhibitor of topoisomerase II rather than a poison. Interestingly, co-treatment of AK-I-190 with etoposide reduced linear DNA formation compared to samples treated with etoposide alone in the cleavage complex assay. This demonstrates that AK-I-190 can be beneficially used in combination with etoposide for reducing genotoxicity while maintaining the efficacy of inhibiting topoisomerase activity. As a DNA intercalating catalytic inhibitor of topoisomerase II, AK-I-190 showed potent anticancer activity through G1 arrest and apoptosis induction. Moreover, AK-I-190 enhanced anti-proliferative activity of paclitaxel, a clinically using chemotherapeutic agent in CRPC.
AR independence in PCa is an important issue that determines the effectiveness of PCa treatment. At the beginning of disease, PCa is manageable, as it is sensitive to androgen-deprivation therapy. However, in a considerable proportion of PCa cases, disease progression with or without prostate serum antigen increase is observed after androgen-deprivation therapy
[16]. These castration-resistant characteristics are closely correlated with metastasis and worse clinical outcome
[4]. Therefore, developing better therapeutic options for CRPC treatment is critical.
Topoisomerase II is an enzyme that is essential for diverse cellular processes, such as transcription, replication, chromosomal condensation and segregation. As topoisomerase II is indispensable in proliferative cells, it has been regarded as an effective therapeutic target for the treatment of solid tumors for decades. Topoisomerase II inhibitors fall into two categories: poisons and catalytic inhibitors
[17][18]. Topoisomerase II poisons act at the stage of DNA cleavage. This type of inhibitor effectively kills cancer cells by trapping the DNA-enzyme complex and blocking DNA re-ligation and enzyme release
[18]. Most of the clinically used topoisomerase II inhibitors are poisons. Despite their highly potent anticancer activity, the use of these compounds has limitations because of their mechanism of action. Topoisomerase poisons stabilize the DNA-enzyme complex, which leads to DNA toxicity. Previous studies have reported that the genotoxicity induced by topoisomerase II poisons causes serious side effects, such as secondary malignancy
[19].
A role of topoisomerase II in PCa has been reported. Amplification of the
TOP2A gene, which encodes topoisomerase II, was shown to correlate with Gleason score and hormone resistance in PCa
[20][21]. Multivariate analysis of PCa cases with systemic progression or death within 5 years after radical retropublic prostatectomy suggested several predictive factors including
TOP2A amplification
[22]. These previous studies indicated that topoisomerase II is not only a predictive factor, but also a promising therapeutic target in PCa. A topoisomerase IIβ–cleavage complex stabilized by topoisomerase II poisons was reported as a possible cause of oncogenic gene fusions, such as
TMPRSS2-ERG [23][24][25].
TMPRSS2-ERG is one of the most common genomic alterations in PCa and is detected in up to 60% of PCa cases
[22][26]. Considering that topoisomerase II inhibitors hardly possess isoform-selectivity between topoisomerase IIα and topoisomerase IIβ, topoisomerase II inhibition without the generation of unwanted DNA double-strand breaks is a critical factor in PCa treatment.
To overcome the drawbacks in the clinical use of topoisomerase II poisons, numerous studies have been conducted to develop catalytic inhibitors with potent activities. Topoisomerase II catalytic inhibitors act at the other steps of the topoisomerase II catalytic cycle, except for the step of DNA-enzyme complex stabilization
[13][27][28][29]. The most important side effect of a topoisomerase II poison is the DNA truncation that is induced by its mechanism of action. We confirmed that AK-I-190 did not induce DNA fragmentation in short-term treatment using the alkaline comet assay. Although DNA fragmentation is a marker of cellular death, it also represents generation of poison-induced DNA truncation under short-term treating condition. We then evaluated the DNA interaction with AK-I-190 by titrating AK-I-190 into ct-DNA, which resulted in induction of hyperchromicity in DNA absorbance spectra. In addition, AK-I-190 replaced EtBr, a DNA intercalating agent, indicating that AK-I-190 interacted with DNA in an intercalating manner. Molecular docking studies showed that AK-I-190 fits into DNA between base pairs and generates additional hydrogen bonds with phosphate groups in the two different chains of DNA. The anticancer activity of AK-I-190 in PCa was also confirmed. AK-I-190 induced G1 arrest and stimulated apoptosis in a dose- and time-dependent manner. Moreover, AK-I-190 combined with paclitaxel, a clinically used anticancer agent, inhibited CRPC cancer cell growth to a greater extent than the single treatments.
The targeting of additional or alternative therapeutic molecular targets can be a solution to improve treatment of patients in CRPC treatment. While topoisomerase II inhibitors have been used for PCa patients, there have been limitations in their clinical use because of their mechanism in stabilizing the DNA-enzyme cleavage complex, producing undesirable truncated DNA. Our results suggest that AK-I-190 may suppress AR-independent PCa cell growth by its potent inhibitory activity against topoisomerase II without genotoxicity. Though the results of this study suggest topoisomerase II inhibition as a realistic alternative for CRPC treatment, the molecular mechanism of how topoisomerase II inhibition affects AR-independent growth of CRPC cells has not been defined. In addition to our results, further studies are needed to elucidate the molecular relationship between topoisomerase II and CRPC.
 +1 credit
+1 credit