 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marco Crescenzi | + 1718 word(s) | 1718 | 2021-10-19 05:10:30 |
Video Upload Options
Terminally differentiated cells are classically defined as specialized cells that have irreversibly lost their ability to proliferate (postmitotic state). Skeletal muscle myotubes are a model system to study terminal differentiation, more amenable than other terminally differentiated histotypes to experimental investigation. Arguably, the fundamental mechanisms underlying the postmitotic state should be shared by most TD cell types.
1. Introduction
Terminally Differentiated (TD) cells are classically defined as specialized cells that have irreversibly lost their ability to proliferate (postmitotic state). This definition, however, is based on the indeterminate notion of “specialization” and on the absence of evidence of proliferation. Both pillars rest on soft ground. We do not know how to objectively measure specialization and what degree of this property, if any, entails terminal differentiation. As to the second pillar, the lack of evidence of proliferation cannot exclude that cells might divide under rare or special conditions. As a relevant example, adult cardiomyocytes, long considered postmitotic, are now established as being endowed with a limited but definite proliferative potential[1]. Indeed, there is ample evidence that at least the cell cycle—or even proliferation—can be reactivated in nearly any cell type, in natural or experimental conditions, and that the postmitotic state can no longer be considered irreversible.
However defined, TD cells, if belonging to tissues with limited or absent renewal, must live as long as their organism itself. This generates the evolutionary problem of ensuring their long-term survival through especially efficient maintenance and repair mechanisms. In addition, they represent a biological mystery, in that we have a limited understanding of the molecular mechanisms that trigger permanent exit from the cell cycle, of what locks the cells in the postmitotic state, and why such a state is so common in mammals and other classes of vertebrates.
Skeletal muscle myotubes are readily generated and easy to cultivate and manipulate in vitro, while the molecular details of their differentiation are understood in depth[2]. For these reasons, they constitute a time-honored model in studies of terminal differentiation. Indeed, mammalian skeletal muscle fibers are excellent examples of postmitotic cells, as under natural conditions they virtually never reenter the cell cycle. Scientists have generally investigated the postmitotic state of TD cells with two aims. On one side, they wish to understand the molecular mechanisms underpinning the decision to abandon proliferation and what makes this choice normally permanent. In doing so, they hope to penetrate the deep significance of the postmitotic state, and its evolutionary advantages and drawbacks. On the other side, they wish to discover how to induce TD cells to proliferate in a controlled, safe, and reversible fashion. Possessing such ability would offer great opportunities to regenerative medicine. It would be invaluable to replace cells lost to diseases or injuries of organs incapable of self-repair through parenchymal cell proliferation. Two general strategies can be envisioned. In ex vivo approaches, healthy TD cells, explanted from a damaged organ and expanded in vitro, would be then transplanted back to replace lost cells. A second possibility is exploiting similar methods for direct, in vivo tissue repair. Reactivation of the cell cycle in TD cells is to be regarded as an approach opposite but complementary to the mastery of stem cells for similar purposes.
Here[3], we review the history and the current state of the efforts to induce TD skeletal muscle cells to reenter the cell cycle and proliferate.
2. The Postmitotic State in Myotubes
The postmitotic state has long been regarded as an attribute of TD cells that have ceased dividing and cannot be recalled into the cell cycle. This definition suggested that such cells are permanently confined in G0 phase. Indeed, they do not synthesize DNA in response to any growth factors, nor to the forced expression of a variety of genes that are powerful mitogenic stimulators in non-TD cells[4]. This static view was initially challenged by the observation that myotubes stimulated with serum or individual growth factors re-express the early cell cycle gene c-Myc[5]. Subsequent studies investigated the control of the cell cycle in postmitotic myotubes in further detail. It was shown that these cells can be readily brought into G1 phase by growth factor stimulation[4]. In fact, the initial transcriptional responses to serum of reversibly quiescent myoblasts and myotubes are indistinguishable, comprising the expression of cell cycle genes such as Fos, Jun, Myc, Id1, and Cyclin D1. However, myotubes display no further response, beyond the expression of cyclin D1, leading to the postulation of a mid-G1 block that prevented these cells from progressing into S phase[4] (Figure 1). Interestingly, growth factor stimulation, though partially reactivating the cell cycle, did not suppress the expression of muscle-specific genes[4][5].
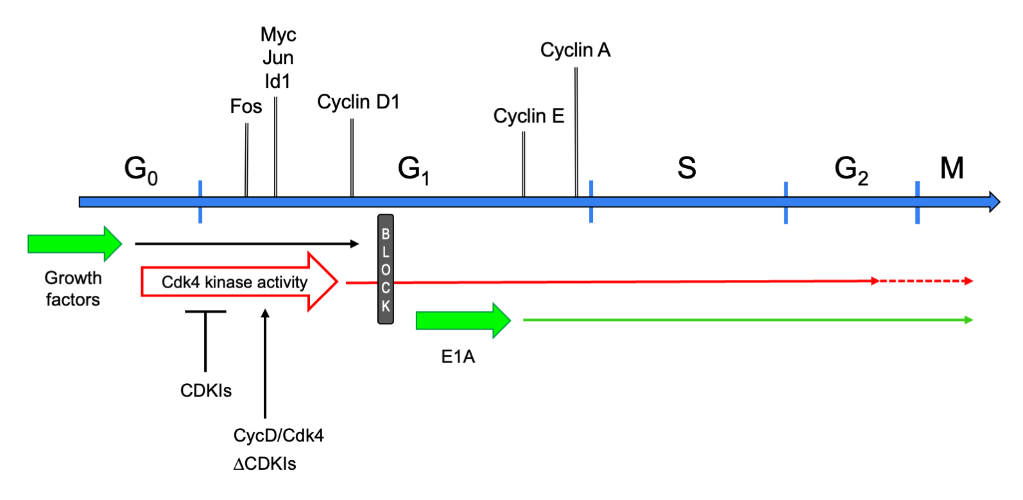 Figure 1. Schematic of the cell cycle in myotubes. Cell cycle phases are graphed as a linear succession. Above the cell cycle line, marker genes are shown at the approximate time point when they are first expressed or upregulated, when reentering the cell cycle from G0. Below the cell cycle line, the effects of several cell cycle-reactivating triggers are presented. Upon growth factor stimulation, TD myotubes exit G0 phase, enter G1, and progress up to the mid-G1 block, which they cannot pass. Expression of E1A makes myotubes jump from G0 to the G1-S boundary. They promptly induce expression of cyclin E and -A, and progress into and beyond M phase. Cyclin D/Cdk4 overexpression (CycD/Cdk4) or CDKI depletion (ΔCDKIs) activates the Cdk4 kinase, allowing myotubes to reach S-G2 phase (CycD/Cdk4) or M phase (ΔCDKIs).
Figure 1. Schematic of the cell cycle in myotubes. Cell cycle phases are graphed as a linear succession. Above the cell cycle line, marker genes are shown at the approximate time point when they are first expressed or upregulated, when reentering the cell cycle from G0. Below the cell cycle line, the effects of several cell cycle-reactivating triggers are presented. Upon growth factor stimulation, TD myotubes exit G0 phase, enter G1, and progress up to the mid-G1 block, which they cannot pass. Expression of E1A makes myotubes jump from G0 to the G1-S boundary. They promptly induce expression of cyclin E and -A, and progress into and beyond M phase. Cyclin D/Cdk4 overexpression (CycD/Cdk4) or CDKI depletion (ΔCDKIs) activates the Cdk4 kinase, allowing myotubes to reach S-G2 phase (CycD/Cdk4) or M phase (ΔCDKIs).
3. Maintenance of the Postmitotic State
It is questionable whether any experimental manipulations, aimed directly at the core cell cycle machinery, allows sustained proliferation of cells derived from myotubes. In fact, it has been described that, in many instances, DNA replication in the reactivated myonuclei—irrespective of their belonging to mono- or multinucleated cells—is incomplete and entails heavy DNA damage[6]. Indeed, it has been proposed that such inability to fully replicate DNA is shared by most TD cells[6]. It has been shown that, in myotubes, incomplete DNA replication is due in part to a defective deoxynucleotide triphosphate (dNTP) pool that limits DNA synthesis. In turn, the deficiency of the dNTP pool is caused by the differentiation-dependent, cell cycle-resistant suppression of genes encoding critical synthetic enzymes, most crucially Thymidine kinase 1 (TK1). However, restoring the dNTP pool allows only partial extension of DNA synthesis, which never reaches completion[6].
Many, but not all, cell cycle genes are silenced in myotubes[4] and this is certainly part of the mechanisms preventing the proliferation of TD cells. The di- or trimethylation of histone H3 lysine 27 (H3K27Me2/3) at these genes has been proposed as one important keeper of the postmitotic state. Indeed, many cell cycle genes acquire the repressive H3K27Me2/3 mark and are silenced during skeletal muscle differentiation. At least some of these genes are also repressed in quiescent fibroblasts, but they do not acquire H3K27Me2/3. Thus, this mark is somehow associated with permanent exit from the cell cycle[7]. Importantly, the depletion of pRb in myotubes shows that its continuing presence is required for the maintenance of H3K27Me2/3 at several genes[7] (Figure 2), adding to the crucial relevance of pRb in the establishment and conservation of the postmitotic state. Interestingly, the Cyclin D1 gene acquires H3K27Me2/3 in myotubes, but in a non-pRb-dependent fashion, probably through the involvement of polycomb group complexes[7].
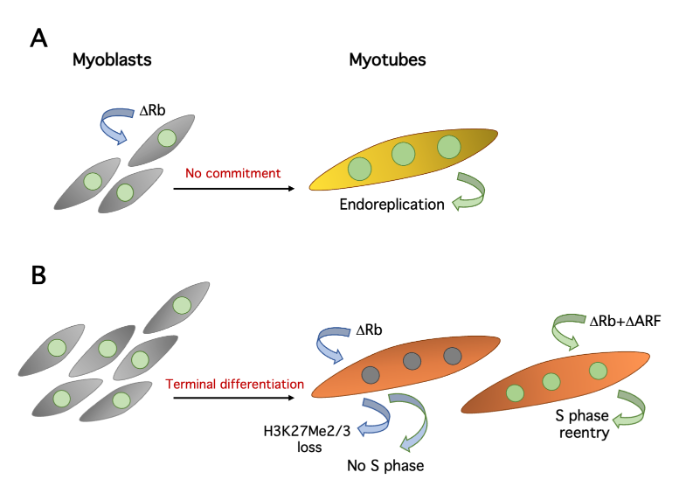
Figure 2. Effects of pRb suppression in primary myoblasts and myotubes. A. Deletion of Rb in myobasts allows defective myotube differentiation without the preceding commitment step, resulting in repeated cycles of endoreplication (large nuclei). B. Rb deletion alone causes loss of H3K27Me2/3 on several cell cycle genes, but rarely triggers S phase. Complementary depletions of pRb and ARF initiate DNA replication.
However, the methylation of H3K27 cannot wholly explain the robustness of the postmitotic state, as most cell cycle genes are readily reexpressed, and presumably lose H3K27Me2/3[7], following a variety of treatments that reactivate the cell cycle in myotubes[8][9][10].
Altogether, these finding might suggest that TD cells are characterized by obstacles to full DNA replication that lie beyond cell cycle control and pertain to differentiation itself. It is still unclear which changes define the postmitotic state and determine its fundamental attributes.
4. Concluding Remarks
Inducing proliferation of myotube-derived cells is still an open problem. Remarkably, however, in the last few years, virtually no new reports have been published on this issue, as if it was considered solved. In our view, this is not the case.
4.1. Lack of Molecular Understanding
In the first place, none of the available methods to induce myotube proliferation is efficient or readily reproducible. However, even if they were, we would still lack a molecu- lar understanding of what constitutes the postmitotic state. Evidence accumulated in the last sixty years shows that TD cells enter a state of permanent proliferation arrest that is qualitatively different from the stances taken by temporarily or permanently nonproliferat- ing cells (e.g., quiescence and senescence). TD cells do not respond to growth factors with proliferation. If forced into the cell cycle, they suppress their differentiation program. When they reenter S phase, TD cells often face obstacles of unknown nature in completing DNA replication. These features require explanations. Terminal differentiation is an unsolved enigma connected with other complex biological problems, such as regeneration, cancer, cell senescence, and organismal aging. Understanding it would shed considerable light on a vast expanse of biology. Skeletal muscle myotubes are a model system to study terminal differentiation, more amenable than other TD histotypes to experimental investigation. Arguably, the fundamental mechanisms underlying the postmitotic state should be shared by most TD cell types.
4.2. Therapeutic Strategies
From a practical standpoint, therapeutic applications are still far into the future. While the skeletal muscle has considerable regenerative capacity, other tissues and organs whose parenchymas are composed of TD cells do not. Examples include the nervous system, sensory organs, the heart (whose cardiomyocyte proliferating capacity is very limited), and endocrine glands. Again, then, the myotube is a model system for TD cell types more difficult to manipulate experimentally. If we succeed in making them proliferate, then very possibly we will be able to do the same with more therapeutically significant target cells. The next challenge will be converting such raw ability into practicable therapeutics, but that is a story yet to be written.
References
- Jaslyn Johnson; Sadia Mohsin; Steven Houser; Cardiomyocyte Proliferation as a Source of New Myocyte Development in the Adult Heart. International Journal of Molecular Sciences 2021, 22, 7764, 10.3390/ijms22157764.
- Hasan A Asfour; Mohammed Z Allouh; Raed S Said; Myogenic regulatory factors: The orchestrators of myogenesis after 30 years of discovery. Experimental Biology and Medicine 2018, 243, 118-128, 10.1177/1535370217749494.
- Deborah Pajalunga; Marco Crescenzi; Restoring the Cell Cycle and Proliferation Competence in Terminally Differentiated Skeletal Muscle Myotubes. Cells 2021, 10, 2753, 10.3390/cells10102753.
- Marianne Tiainen; Deborah Pajalunga; Flavia Ferrantelli; Silvia Soddu; G Salvatori; A Sacchi; Marco Crescenzi; Terminally differentiated skeletal myotubes are not confined to G0 but can enter G1 upon growth factor stimulation.. Cell growth & differentiation : the molecular biology journal of the American Association for Cancer Research 1996, 7, 1039-1050.
- T Endo; B Nadal-Ginard; Transcriptional and posttranscriptional control of c-myc during myogenesis: its mRNA remains inducible in differentiated cells and does not suppress the differentiated phenotype. Molecular and Cellular Biology 1986, 6, 1412-1421, 10.1128/mcb.6.5.1412-1421.1986.
- Deborah Pajalunga; Eleonora M. R. Puggioni; Alessia Mazzola; Valentina Leva; Alessandra Montecucco; Marco Crescenzi; DNA Replication Is Intrinsically Hindered in Terminally Differentiated Myotubes. PLoS ONE 2010, 5, e11559-e11559, 10.1371/journal.pone.0011559.
- Alexandre Blais; Chris J.C. van Oevelen; Raphaël Margueron; Diego Acosta-Alvear; Brian David Dynlacht; Retinoblastoma tumor suppressor protein–dependent methylation of histone H3 lysine 27 is associated with irreversible cell cycle exit. Journal of Cell Biology 2007, 179, 1399-1412, 10.1083/jcb.200705051.
- M Tiainen; D Spitkovsky; P Jansen-Dürr; A Sacchi; M Crescenzi; Expression of E1A in terminally differentiated muscle cells reactivates the cell cycle and suppresses tissue-specific genes by separable mechanisms. Molecular and Cellular Biology 1996, 16, 5302-5312, 10.1128/mcb.16.10.5302.
- Lucia Latella; Alessandra Sacco; Deborah Pajalunga; Marianne Tiainen; Daniela Macera; Marco D'Angelo; Angelina Felici; Ada Sacchi; Marco Crescenzi; Reconstitution of Cyclin D1-Associated Kinase Activity Drives Terminally Differentiated Cells into the Cell Cycle. Molecular and Cellular Biology 2001, 21, 5631-5643, 10.1128/mcb.21.16.5631-5643.2001.
- Nikolaos P. Mastroyiannopoulos; Paschalis Nicolaou; Mustafa Anayasa; James B. Uney; Leonidas A. Phylactou; Down-Regulation of Myogenin Can Reverse Terminal Muscle Cell Differentiation. PLoS ONE 2012, 7, e29896, 10.1371/journal.pone.0029896.
- Nikolaos P. Mastroyiannopoulos; Paschalis Nicolaou; Mustafa Anayasa; James B. Uney; Leonidas A. Phylactou; Down-Regulation of Myogenin Can Reverse Terminal Muscle Cell Differentiation. PLoS ONE 2012, 7, e29896, 10.1371/journal.pone.0029896.

