Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Tien-Jyun Chang | + 2611 word(s) | 2611 | 2021-10-15 07:42:13 | | | |
| 2 | Vivi Li | Meta information modification | 2611 | 2021-10-18 05:15:14 | | | | |
| 3 | Dean Liu | Meta information modification | 2611 | 2021-11-11 04:45:36 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Chang, T. Aldehyde Dehydrogenase. Encyclopedia. Available online: https://encyclopedia.pub/entry/15068 (accessed on 28 June 2026).
Chang T. Aldehyde Dehydrogenase. Encyclopedia. Available at: https://encyclopedia.pub/entry/15068. Accessed June 28, 2026.
Chang, Tien-Jyun. "Aldehyde Dehydrogenase" Encyclopedia, https://encyclopedia.pub/entry/15068 (accessed June 28, 2026).
Chang, T. (2021, October 15). Aldehyde Dehydrogenase. In Encyclopedia. https://encyclopedia.pub/entry/15068
Chang, Tien-Jyun. "Aldehyde Dehydrogenase." Encyclopedia. Web. 15 October, 2021.
Copy Citation
Chronic hyperglycemia and hyperlipidemia hamper beta cell function, leading to glucolipotoxicity. Mitochondrial aldehyde dehydrogenase 2 (ALDH2) detoxifies reactive aldehydes, such as methylglyoxal (MG) and 4-hydroxynonenal (4-HNE), derived from glucose and lipids, respectively.
aldehyde dehydrogenase 2 (ALDH2)
glucolipotoxicity
beta cell function
Alda-1
1. Introduction
Insulin resistance and progressive pancreatic beta cell dysfunction are the main features of type 2 diabetes mellitus, most likely owing to a vicious cycle involving the accumulation of toxic aldehydes and relative oxidative stress [1]. It is well known that chronic hyperglycemia and hyperlipidemia, also known as glucolipotoxicity, are harmful to beta cell function. Under glucolipotoxicity, the fine balance between the levels of pro-oxidants and antioxidants in pancreatic beta cells is disturbed, which leads to a chronic oxidative stress that subsequently contributes to impaired glucose-stimulated insulin secretion (GSIS) [2][3]. Pancreatic beta cells are particularly susceptible to oxidative stress, which contributes to beta cell dysfunction and cell death [4]. The decreases in antioxidant defense, hyperglycemia, inflammation, and obesity contribute to the accumulation of toxic aldehydes, including glyoxal, methylglyoxal (MG), glycolaldehyde, and 4-hydroxynonenal (4-HNE), among others [5]. The elevated levels of toxic aldehydes can damage carbohydrates, amino acids, and lipids, leading to the formation of reactive carbonyl compounds (RCCs), which further react with macromolecules and yield advanced glycation end products (AGE) or advanced lipoxidation end products (ALE). AGEs bind to the cell surface receptor for the advanced glycation end products (RAGE) and induce the phosphorylation of PKC and subsequent activation of NADPH oxidase, thereby leading to theexcessive intracellular reactive oxygen species (ROS) formation [5] and the activation of key transcriptional factors, such as NFκB, AP1, Nrf2, HSF1, PDX1 and FOXO1 [6]. The alteration of transcriptional factors by toxic aldehydes modifies the gene expression of proinflammatory cytokines, detoxifying genes, heat shock proteins (HSP), insulin gene expression, and beta cell proliferation [6].
Cohen et al. studied the effects of 4-HNE on the rat islets of Langerhans and the rat insulinoma INS-1E beta cell line and demonstrated that exogenously added, dose-dependent 4-HNE induced apoptosis and cell death [7]. MG is an intracellularly formed α-ketoaldehyde that is involved in the formation of AGEs. The accumulation of MG contributes to glucotoxicity and mediates beta cell apoptosis [8].
Aldehyde dehydrogenase 2 (ALDH2) is located in the mitochondria, where it plays a major role in acetaldehyde detoxification in humans and the detoxification of ROS-generated aldehyde adducts [9]. ALDH2 transgenic mice showed an enhanced acetaldehyde detoxification following chronic alcohol intake, leading to an improved whole-body glucose tolerance, cardiac glucose uptake and insulin signaling at the receptor and post-receptor levels [10]. The ALDH2 activator, Alda-1, significantly accelerated adipocyte differentiation in 3T3-L1 cells through the regulation of PPARγ transcriptional activity [11]. Furthermore, the administration of Alda-1 decreased 4-HNE concentration [12] and attenuated cardiac [12][13] and renal damage [14] induced by ischemia-reperfusion injury through increased oxidative stress. Therefore, it is reasonable to hypothesize that the administration of an ALDH2 activator can ameliorate beta cell dysfunction and apoptosis induced by glucolipotoxicity. In this study, we showed that the prototype ALDH2 activator (Alda-1) significantly enhanced insulin secretion by improving the mitochondrial function of the beta cells. Moreover, Alda-1 ameliorated either MG- or 4-HNE-induced beta cell apoptosis by alleviating the production of superoxide from the mitochondria and the cytoplasm, subsequently leading to improved mitochondrial function, decreasing beta cell apoptosis and death.
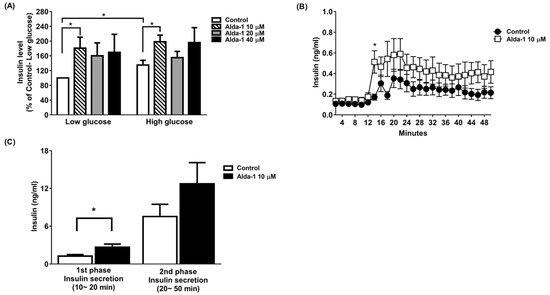
2. ALDH2 Activator Enhanced GSIS
We tested different doses (10, 20, and 40 μM) of ALDH2 activator Alda-1 on the enhancement of GSIS, and found that Alda-1 at 10 μM potentiated static insulin secretion under both low (3.3 mM) and high (16.7 mM) glucose concentrations in cultured MIN6 cells (Figure 1A).
Figure 1. Alda-1 promotes insulin secretion in MIN6 pancreatic beta cells. (A) Insulin secretory response in MIN6 cells exposed to different doses of Alda-1 under low (3.3 mM) and high glucose (16.7 mM) concentrations (n = 3/group). (B) Glucose-stimulated insulin secretion in ex vivo perifused islets in the absence and presence of 10 μM Alda-1. (C) First and second phase insulin secretions were measured from the islet perifusion study (details in the Method section). (n = 14 in the control group, n = 15 in the Alda-1 10 μM group). Data are presented as mean ± SEM * p < 0.05 versus the control group.
To further study GSIS in primary islets, an ex vivo primary islet perifusion study was performed. Alda-1 at 10 μM significantly promoted GSIS at 4 min (i.e., 14 min during the perifusion study) after switching the perifusates from a basal to a high glucose concentration (16.7 mM), and continuously increased insulin secretion during the course of the islet perifusion study (Figure 1B). The first phase (0–10 min during high glucose perifusion) and second phase (10–40 min during high glucose perifusion) of insulin secretion were analyzed, and Alda-1 significantly potentiated first-phase insulin secretion (Figure 1C).
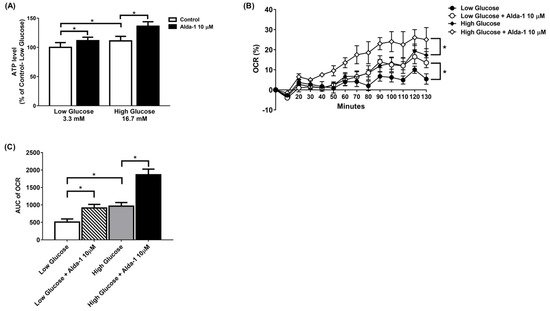
3. ALDH2 Activator Improved Mitochondrial Function of Pancreatic Beta Cells
The ALDH2 activator Alda-1 significantly increased the intracellular ATP content in MIN6 cells cultured under both low and high glucose conditions. (Figure 2A). To further explore the changes in the mitochondrial function of the MIN6 cells under different culture conditions, we employed a Seahorse XF analyzer to measure the OCR. The data showed that Alda-1 increased the OCR for both low and high glucose incubation conditions, which was significant for p-for-trend analyses (Figure 2B). We also compared the AUC of the OCR for MIN6 cells under different conditions. As shown in Figure 2C, Alda-1 treatment significantly increased the AUC of OCR in MIN6 cells under both low and high glucose conditions (Figure 2C). Notably, the effect of Alda-1 on mitochondrial function was independent of ambient glucose concentrations (Figure 2B,C).
Figure 2. Alda-1 improves mitochondrial function. (A) ATP level in response to 10 μM Alda-1 in MIN6 cells under low (3.3 mM) and high glucose (16.7 mM) concentrations. (B) Effect of Alda-1 on oxygen consumption rate (OCR) in MIN6 beta cells under low (3.3 mM) and high glucose (16.7 mM) concentrations. (C) The area under curves (AUC) of the OCR. Data are presented as mean ± SEM of three independent experiments (n = 3 per group). * p < 0.05 versus the control group with p-for-trend analysis and Student’s t-test, respectively.
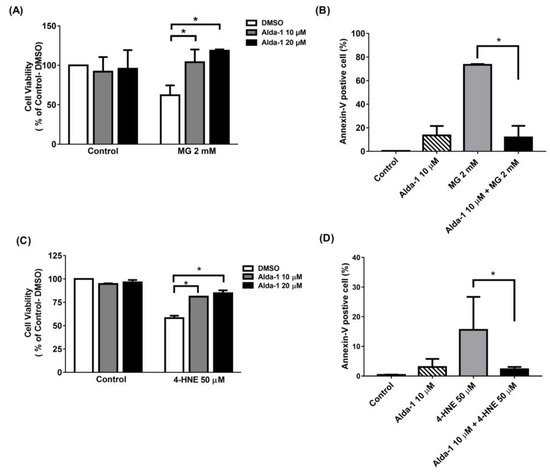
4. Alda-1 Rescued MIN6 Cells from MG- and 4-HNE- Induced Beta Cell Death and Apoptosis
To investigate the effect of the ALDH2 activator on the improvement of beta cell survival, we measured MIN6 cell survival upon MG and 4-HNE exposure, mimicking glucolipotoxicity conditions. We found that Alda-1 dose-dependently rescued cell survival when MIN6 cells were treated with 2 mM MG (Figure 3A) or 50 μM 4-HNE (Figure 3C). To understand the mechanism underlying cell survival, we used flow cytometry with Annexin V to study the potential effect of Alda-1 on cell apoptosis and/or necrosis. As shown in Figure 3B,D, the pretreatment of MIN6 cells with Alda-1 significantly decreased MG- and 4-HNE-induced beta cell apoptosis.
Figure 3. Alda-1 rescues the cell death induced by glucotoxicity and lipotoxicity via anti-apoptotic effect. (A) Cell viability in the control and Alda-1-treated MIN6 cells in the absence and presence of 2 mM methylglyoxal (MG). (B) The percentages of Annexin-V-positive and AAD-7-negative MIN6 cells treated with or without Alda-1 in the absence and presence of 2 mM methylglyoxal (MG) for 24 h. (C) Cell viability in control and Alda-1-treated MIN6 cells in the absence and presence of 50 μM 4-Hydroxynonenal (4-HNE). (D) The percentages of the Annexin-V-positive and AAD-7-negative MIN6 cells treated with or without Alda-1 in the absence and presence of 50 μM 4-HNE for 24 h. Data are presented as mean ± SEM of three independent experiments (n = 3 per group). * p < 0.05.
5. Discussion
In this study, we found that Alda-1, an ALDH2 activator, potentiated insulin secretion in both beta cells and mouse primary islets by improving mitochondrial function. On the other hand, Alda-1 also ameliorated the harmful effects of glucolipotoxicity on beta cells by improving mitochondrial function and reducing ROS production, as well as the apoptosis of beta cells. To the best of our knowledge, this was the first study to demonstrate the rescuing glucolipotoxicity effects of the ALDH2 activator in pancreatic beta cells.
The prevalence of type 2 diabetes mellitus has increased worldwide [15]. Insulin resistance and progressive pancreatic beta cell failure are the main features of type 2 diabetes mellitus [1]. The progressive loss of beta cell mass and the progressive decline in beta cell function are the main pathogeneses leading to the progression of type 2 diabetes [16]. The targets for the treatment of diabetes mellitus remain unsatisfactory despite several classes of anti-diabetic agents used in the clinical setting. Most types of therapy eventually fail as type 2 diabetes is a progressive disorder. Therefore, there is still an unmet medical need for the sustained and effective treatment of type 2 diabetes. High glucose concentration increases the cytosolic ATP level, which induces the closure of KATP channels and results in cell membrane depolarization, followed by the opening of the voltage-dependent calcium channel. Subsequently, this leads to a Ca2+ influx in the cells with increased [Ca2+]i, thereby promoting the exocytosis of insulin-containing granules [17]. The triggering pathway is essential for the first phase of insulin secretion [17]. In the process of GSIS, the glycolytic flux is tightly coupled to increased mitochondrial oxidative activity, leading to the increased production of ROS [18]. ALDH2 is a nuclear-coded aldehyde oxidase that is localized in the mitochondrial matrix. Many studies confirmed that ALDH2 can decompose the acetaldehyde metabolite 4-HNE and mitigate oxidative damage to the cells induced by acetaldehyde and its metabolites [19]. A recent study reported that fibroblasts of a patient with Alzheimer’s disease (AD) had approximately 25% ALDH2 activity relative to the fibroblasts of a healthy subject. The AD-derived fibroblasts increased mitochondrial ROS production, reduced ATP levels, reduced mitochondrial respiration (OXPHOS), and caused a shift towards glycolysis (ECAR) relative to the fibroblasts derived from healthy subjects. All of the above defects observed in AD-derived fibroblasts were significantly corrected with Alda-1 treatment [20]. Another study also showed that the impairment of ALDH2 accelerated the acquisition of a premature senescent phenotype in endothelial cells, a change likely to be associated with the observed reduction in mitochondrial respiration and its reserved capacity [21]. In this study, the ALDH2 activator Alda-1 potentiated insulin secretion in MIN6 cells and first-phase insulin secretion in primary islets by improving mitochondrial function as indicated by an increase in the intracellular ATP concentration and oxygen consumption rate. The improved mitochondrial function may be caused by the elimination of ROS and subsequent enhancement of insulin secretion via the increased intracellular ATP/ADP ratio. The potentiation effect of Alda-1 on insulin secretion and the intracellular ATP concentrations were abolished in Aldh2-knockdown MIN6 cells, indicating that the effect of Alda-1 was mediated through the activation of ALDH2. More interestingly, the intracellular ATP concentrations under conditions of high glucose concentrations were similar to those in Aldh2-knockdown MIN6 cells cultured under high glucose concentrations together with Alda-1, suggested that the effect of improving mitochondrial function was caused by the activation of ALDH2 independent of glucose concentration.
The long-term exposure to high concentrations of glucose and non-esterified free fatty acid (NEFA) in beta cells altered membrane fluidity, protein palmitoylation, and ceramide production, which resulted in mitochondrial dysfunction, endoplasmic reticulum (ER) stress, autophagy, and apoptosis [22][23][24][25]. Arachidonic acid and linoleic acid were subjected to peroxidation, resulting in the generation of 4-HNE, which induced apoptosis and cell death in terms of lipotoxicity [26]. ALDH could oxidize 4-HNE to 4-hydroxy-2-nonenoic acid (HNA), which was one of the three major detoxification pathways for converting 4-HNE to a less reactive chemical species [27]. ALDH2, a member of the ALDH family, was exclusively located in the mitochondria [27]. The prototype of the ALDH2 activator, Alda-1, activated the wild type enzyme and restored the activity of the ALDH2*2 mutant enzyme by acting as a structural chaperone [28]. In a recent study, the activation of ALDH2 prevented the cardiac-arrest-induced death of cardiomyocytes from 4-HNE-induced mitochondrial ROS production and the subsequent mitochondrial damage and cell apoptosis [29]. In this study, we demonstrated that Alda-1 ameliorated 4-HNE-induced beta cell death, apoptosis, and mitochondrial, as well as cytoplasmic ROS levels. Moreover, Alda-1 significantly restored the 4-HNE-induced reduction in intracellular ATP concentration in a time-dependent manner. Finally, we also showed that the pretreatment with Alda-1 decreased the expression of apoptotic molecules, such as cleaved PARP, cleaved caspase 3, and Bax. However, the expression of anti-apoptotic molecules, such as MCL-1, Bcl-2, and p-Akt, was not affected by Alda-1. Consistently, the alleviating effect of Alda-1 on beta cell death was abrogated in the Aldh2-knockdown MIN6 cells, which validated the effect of Alda-1 on beta cell viability through the activation of ALDH2.
Chronic hyperglycemia leads to the formation of AGE by promoting the non-enzymatic glycation of endogenous proteins, lipids and nucleic acids [30]. MG is an intracellularly formed α-ketoaldehyde, which is an essential source of intracellular AGEs. It is reported that MG suppresses the oxygen consumption rate and decreases intracellular ATP levels in RINm5F beta cells [31]. Several reports also demonstrated that MG or glyoxal reduces the mitochondrial membrane potential, suppresses the activities of respiratory chain complexes, decreases the ATP production, and elevates the ROS levels in different cells [32][33][34]. Moreover, MG increases the intracellular ROS production and lactate levels and decreases the mitochondrial membrane potential and intracellular ATP levels in SH-SY5Y neuroblastoma cells. The MG-induced depletion of ATP and mitochondrial dysfunction can be prevented by the pretreatment with the carbonyl scavengers aminoguanidine and tenilsetam [33]. Although MG is a substrate of ALDH2 [9], the alleviation of MG-induced beta cell death and apoptosis by ALDH2 activation is not reported. In this study, we showed that Alda-1 ameliorated MG-induced beta cell death, apoptosis (Figure 3B), and mitochondrial, as well as cytoplasmic, ROS production. Moreover, Alda-1 restored the MG-suppressed intracellular ATP concentration, suggesting that the activation of ALDH2 improved mitochondrial function. Furthermore, we demonstrated that the rescue effect of Alda-1 on beta cell viability was abolished in Aldh2 knockdown MIN6 cells, indicating that the effect of Alda-1 on beta cell survival was mediated by the activation of ALDH2.
The high circulating levels of both glucose and free fatty acids are known to induce oxidative stress in beta cells [35][36]. Beta cells are particularly sensitive to oxidative stress due to the low expression levels of antioxidant enzymes [4]. In this study, we demonstrated that Alda-1 not only alleviated MG- and 4-HNE-induced beta cell dysfunction, apoptosis and death, but also ameliorated either palmitate per se or both high glucose- and palmitate-evoked toxic effects. Therefore, the activation of ALDH2 attenuated glucolipotoxicity induced from high glucose and fatty acids, alone and by their toxic byproducts, such as MG and 4-HNE.
Because of the limitation of human islets resources, we only used a beta cell line and primary islets from mice to demonstrate the protective effect of Alda-1 from glucolipotoxicity on beta cells in this study. We will conduct an experiment on human primary islets in the future study. Another limitation of this study was that the MIN6 cells used in the experiments underwent a long-term passage. According to previous observations [37][38], high-passage MIN6 cells lose their ability to secrete insulin in response to glucose, especially with no first-phase insulin secretion and an impaired second-phase GSIS. The phenotypes observed in the high passage MIN6 cells are then similar to patients with an early onset of type 2 diabetes. In our study, we used high-passage MIN6 cells and found a small but significant response of insulin secretion to high glucose (Figure 1A. insulin level in high glucose: 135% of Control-Low glucose, p = 0.042). This finding was compatible with previous reports [37][38]. In this circumstance, the ALDH2 activator Alda-1 still potentiated the insulin secretion of MIN6 cells in both low and high glucose concentrations, suggesting a potential for the treatment of type 2 diabetes subjects.
Pancreatic beta cell failure is pivotal to diabetes development [39][40][41] and the preservation of functional beta cells can change the clinical outcome of diabetes [42][43]. However, none of the current anti-diabetic drugs reversed the progression of beta cell dysfunction and death. In this study, we developed a new strategy of preserving beta cell function and cell viability by activating ALDH2 to detoxify glucolipotoxicity-induced ROS production, decreasing mitochondrial function and subsequent beta cell dysfunction, cell apoptosis and death.
References
- O’Brien, P.J.; Siraki, A.G.; Shangari, N. Aldehyde sources, metabolism, molecular toxicity mechanisms, and possible effects on human health. Crit. Rev. Toxicol. 2005, 35, 609–662.
- Robertson, R.P.; Harmon, J.; Tran, P.O.; Poitout, V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 2004, 53 (Suppl. 1), S119–S124.
- Kohnke, R.; Mei, J.; Park, M.; York, D.A.; Erlanson-Albertsson, C. Fatty acids and glucose in high concentration down-regulates ATP synthase beta-subunit protein expression in INS-1 cells. Nutr. Neurosci. 2007, 10, 273–278.
- Rhodes, C.J. Type 2 diabetes-a matter of beta-cell life and death? Science 2005, 307, 380–384.
- Thallas-Bonke, V.; Thorpe, S.R.; Coughlan, M.T.; Fukami, K.; Yap, F.Y.; Sourris, K.C.; Penfold, S.A.; Bach, L.A.; Cooper, M.E.; Forbes, J.M. Inhibition of NADPH oxidase prevents advanced glycation end product-mediated damage in diabetic nephropathy through a protein kinase C-alpha-dependent pathway. Diabetes 2008, 57, 460–469.
- Jaganjac, M.; Tirosh, O.; Cohen, G.; Sasson, S.; Zarkovic, N. Reactive aldehydes--second messengers of free radicals in diabetes mellitus. Free Radic. Res. 2013, 47 (Suppl. 1), 39–48.
- Cohen, G.; Riahi, Y.; Shamni, O.; Guichardant, M.; Chatgilialoglu, C.; Ferreri, C.; Kaiser, N.; Sasson, S. Role of lipid peroxidation and PPAR-delta in amplifying glucose-stimulated insulin secretion. Diabetes 2011, 60, 2830–2842.
- Sheader, E.A.; Benson, R.S.; Best, L. Cytotoxic action of methylglyoxal on insulin-secreting cells. Biochem. Pharmacol. 2001, 61, 1381–1386.
- Chen, C.H.; Cruz, L.A.; Mochly-Rosen, D. Pharmacological recruitment of aldehyde dehydrogenase 3A1 (ALDH3A1) to assist ALDH2 in acetaldehyde and ethanol metabolism in vivo. Proc. Natl. Acad. Sci. USA 2015, 112, 3074–3079.
- Li, S.Y.; Gilbert, S.A.; Li, Q.; Ren, J. Aldehyde dehydrogenase-2 (ALDH2) ameliorates chronic alcohol ingestion-induced myocardial insulin resistance and endoplasmic reticulum stress. J. Mol. Cell. Cardiol. 2009, 47, 247–255.
- Yu, Y.H.; Liao, P.R.; Guo, C.J.; Chen, C.H.; Mochly-Rosen, D.; Chuang, L.M. PKC-ALDH2 Pathway Plays a Novel Role in Adipocyte Differentiation. PLoS ONE 2016, 11, e0161993.
- Chen, C.H.; Budas, G.R.; Churchill, E.N.; Disatnik, M.H.; Hurley, T.D.; Mochly-Rosen, D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 2008, 321, 1493–1495.
- Gomes, K.M.; Campos, J.C.; Bechara, L.R.; Queliconi, B.; Lima, V.M.; Disatnik, M.H.; Magno, P.; Chen, C.H.; Brum, P.C.; Kowaltowski, A.J.; et al. Aldehyde dehydrogenase 2 activation in heart failure restores mitochondrial function and improves ventricular function and remodelling. Cardiovasc. Res. 2014, 103, 498–508.
- Zhong, Z.; Hu, Q.; Fu, Z.; Wang, R.; Xiong, Y.; Zhang, Y.; Liu, Z.; Wang, Y.; Ye, Q. Increased Expression of Aldehyde Dehydrogenase 2 Reduces Renal Cell Apoptosis during Ischemia/Reperfusion Injury after Hypothermic Machine Perfusion. Artif. Organs 2016, 40, 596–603.
- Mokdad, A.H.; Ford, E.S.; Bowman, B.A.; Dietz, W.H.; Vinicor, F.; Bales, V.S.; Marks, J.S. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. JAMA 2003, 289, 76–79.
- Kahn, S.E. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia 2003, 46, 3–19.
- Rorsman, P.; Renstrom, E. Insulin granule dynamics in pancreatic beta cells. Diabetologia 2003, 46, 1029–1045.
- Sekine, N.; Cirulli, V.; Regazzi, R.; Brown, L.J.; Gine, E.; Tamarit-Rodriguez, J.; Girotti, M.; Marie, S.; MacDonald, M.J.; Wollheim, C.B.; et al. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic beta-cells. Potential role in nutrient sensing. J. Biol. Chem. 1994, 269, 4895–4902.
- Zhang, Y.; Ren, J. ALDH2 in alcoholic heart diseases: Molecular mechanism and clinical implications. Pharmacol. Ther. 2011, 132, 86–95.
- Joshi, A.U.; Van Wassenhove, L.D.; Logas, K.R.; Minhas, P.S.; Andreasson, K.I.; Weinberg, K.I.; Chen, C.H.; Mochly-Rosen, D. Aldehyde dehydrogenase 2 activity and aldehydic load contribute to neuroinflammation and Alzheimer’s disease related pathology. Acta Neuropathol. Commun. 2019, 7, 190.
- Nunemaker, C.S.; Wasserman, D.H.; McGuinness, O.P.; Sweet, I.R.; Teague, J.C.; Satin, L.S. Insulin secretion in the conscious mouse is biphasic and pulsatile. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E523–E529.
- Kharroubi, I.; Ladriere, L.; Cardozo, A.K.; Dogusan, Z.; Cnop, M.; Eizirik, D.L. Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: Role of nuclear factor-kappaB and endoplasmic reticulum stress. Endocrinology 2004, 145, 5087–5096.
- Maedler, K.; Spinas, G.A.; Dyntar, D.; Moritz, W.; Kaiser, N.; Donath, M.Y. Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 2001, 50, 69–76.
- Stein, D.T.; Stevenson, B.E.; Chester, M.W.; Basit, M.; Daniels, M.B.; Turley, S.D.; McGarry, J.D. The insulinotropic potency of fatty acids is influenced profoundly by their chain length and degree of saturation. J. Clin. Investig. 1997, 100, 398–403.
- Elsner, M.; Gehrmann, W.; Lenzen, S. Peroxisome-generated hydrogen peroxide as important mediator of lipotoxicity in insulin-producing cells. Diabetes 2011, 60, 200–208.
- Cohen, G.; Shamni, O.; Avrahami, Y.; Cohen, O.; Broner, E.C.; Filippov-Levy, N.; Chatgilialoglu, C.; Ferreri, C.; Kaiser, N.; Sasson, S. Beta cell response to nutrient overload involves phospholipid remodelling and lipid peroxidation. Diabetologia 2015, 58, 1333–1343.
- Zhong, H.; Yin, H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing on mitochondria. Redox Biol. 2015, 4, 193–199.
- Perez-Miller, S.; Younus, H.; Vanam, R.; Chen, C.H.; Mochly-Rosen, D.; Hurley, T.D. Alda-1 is an agonist and chemical chaperone for the common human aldehyde dehydrogenase 2 variant. Nat. Struct. Mol. Biol. 2010, 17, 159–164.
- Zhang, R.; Liu, B.; Fan, X.; Wang, W.; Xu, T.; Wei, S.; Zheng, W.; Yuan, Q.; Gao, L.; Yin, X.; et al. Aldehyde Dehydrogenase 2 Protects against Post-Cardiac Arrest Myocardial Dysfunction through a Novel Mechanism of Suppressing Mitochondrial Reactive Oxygen Species Production. Front. Pharmacol. 2020, 11, 373.
- Schalkwijk, C.G.; Brouwers, O.; Stehouwer, C.D. Modulation of insulin action by advanced glycation end products: A new player in the field. Horm. Metab. Res. 2008, 40, 614–619.
- Chang, T.J.; Tseng, H.C.; Liu, M.W.; Chang, Y.C.; Hsieh, M.L.; Chuang, L.M. Glucagon-like peptide-1 prevents methylglyoxal-induced apoptosis of beta cells through improving mitochondrial function and suppressing prolonged AMPK activation. Sci. Rep. 2016, 6, 23403.
- Shangari, N.; O’Brien, P.J. The cytotoxic mechanism of glyoxal involves oxidative stress. Biochem. Pharmacol. 2004, 68, 1433–1442.
- De Arriba, S.G.; Stuchbury, G.; Yarin, J.; Burnell, J.; Loske, C.; Munch, G. Methylglyoxal impairs glucose metabolism and leads to energy depletion in neuronal cells--Protection by carbonyl scavengers. Neurobiol. Aging 2007, 28, 1044–1050.
- Wang, H.; Liu, J.; Wu, L. Methylglyoxal-induced mitochondrial dysfunction in vascular smooth muscle cells. Biochem. Pharmacol. 2009, 77, 1709–1716.
- Robertson, R.P. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J. Biol. Chem. 2004, 279, 42351–42354.
- Hasnain, S.Z.; Prins, J.B.; McGuckin, M.A. Oxidative and endoplasmic reticulum stress in beta-cell dysfunction in diabetes. J. Mol. Endocrinol. 2016, 56, R33–R54.
- Cheng, K.; Delghingaro-Augusto, V.; Nolan, C.J.; Turner, N.; Hallahan, N.; Andrikopoulos, S.; Gunton, J.E. High passage MIN6 cells have impaired insulin secretion with impaired glucose and lipid oxidation. PLoS ONE 2012, 7, e40868.
- O’Driscoll, L.; Gammell, P.; McKiernan, E.; Ryan, E.; Jeppesen, P.B.; Rani, S.; Clynes, M. Phenotypic and global gene expression profile changes between low passage and high passage MIN-6 cells. J. Endocrinol. 2006, 191, 665–676.
- Matthews, D.R.; Cull, C.A.; Stratton, I.M.; Holman, R.R.; Turner, R.C. UKPDS 26: Sulphonylurea failure in non-insulin-dependent diabetic patients over six years. UK Prospective Diabetes Study (UKPDS) Group. Diabet. Med. 1998, 15, 297–303.
- Donath, M.Y.; Halban, P.A. Decreased beta-cell mass in diabetes: Significance, mechanisms and therapeutic implications. Diabetologia 2004, 47, 581–589.
- Harrity, T.; Farrelly, D.; Tieman, A.; Chu, C.; Kunselman, L.; Gu, L.; Ponticiello, R.; Cap, M.; Qu, F.; Shao, C.; et al. Muraglitazar, a novel dual (alpha/gamma) peroxisome proliferator-activated receptor activator, improves diabetes and other metabolic abnormalities and preserves beta-cell function in db/db mice. Diabetes 2006, 55, 240–248.
- Defronzo, R.A. Banting Lecture. From the triumvirate to the ominous octet: A new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009, 58, 773–795.
- Leahy, J.L.; Hirsch, I.B.; Peterson, K.A.; Schneider, D. Targeting beta-cell function early in the course of therapy for type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2010, 95, 4206–4216.
More
Information
Subjects:
Others
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
3 times
(View History)
Update Date:
11 Nov 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No