 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Shih-Cheng Li | + 2290 word(s) | 2290 | 2021-09-26 04:21:16 | | | |
| 2 | Conner Chen | Meta information modification | 2290 | 2021-10-09 02:48:05 | | |
Video Upload Options
Zeolites are microporous aluminosilicates with high surface area and crystallinity. They have been widely applied in many different fields, such as gas storage, water treatment, biomass upgrading, and oil refining, because of their strong acidity, excellent catalytic activity, shape selectivity, and hydrothermal stability. In the past decades, one of the most important applications of zeolites is in fluidized catalytic cracking (FCC) in the petrochemical industry, which accounts for more than 95% of the global zeolite catalyst consumption. It is reported that 400 million tons of olefins are produced annually, and about 59% of olefins are produced by FCC units. Light olefins are critical building blocks in the petrochemical industry, and the demand for olefins and their derivatives has continuously increased over the last decade. Therefore, it is important to understand how to improve the catalytic performance of zeolites. Studies have shown that the performance of zeolite catalysts for cracking reactions is determined by various factors, including the porous size and composition, e.g., the Si/Al ratio and the presence of other heteroatoms or extra-framework aluminum (EFAL) species. Since the range of possible combinations of zeolite structures and compositions is exceedingly large, it is highly desirable to understand the effects of zeolite topology and composition on hydrocarbon cracking in order to improve their activity and selectivity to desired products.
1. The Effects of Zeolite Structures on Alkane Cracking
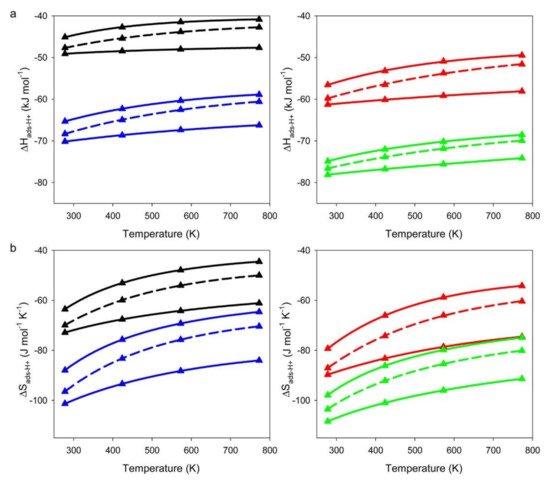
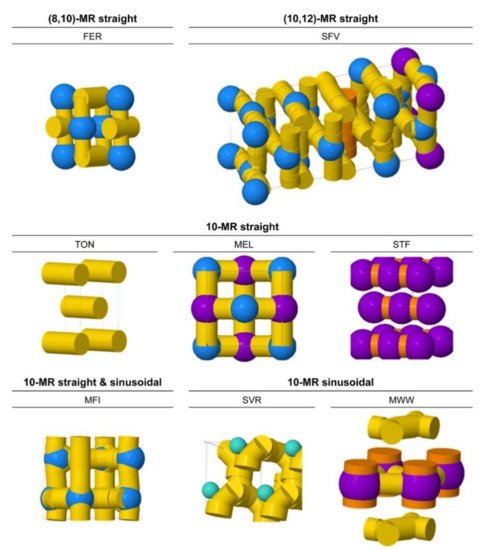
2. The Effects of Mesoporosity on Alkane Cracking
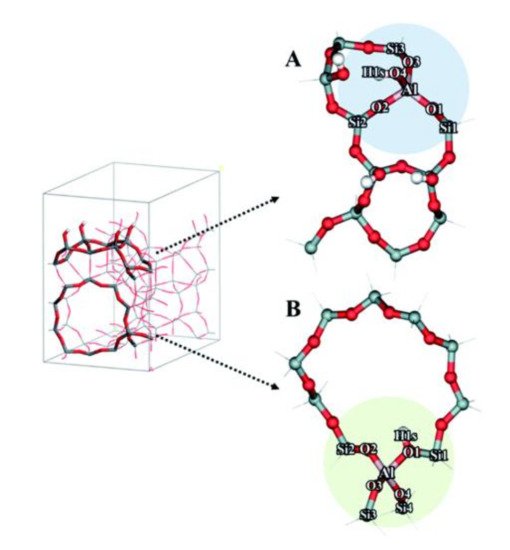
References
- Janda, A.; Vlaisavljevich, B.; Lin, L.-C.; Smit, B.; Bell, A.T. Effects of zeolite structural confinement on adsorption thermodynamics and reaction kinetics for monomolecular cracking and dehydrogenation of n-butane. J. Am. Chem. Soc. 2016, 138, 4739–4756.
- Van der Mynsbrugge, J.; Janda, A.; Sharada, S.M.; Lin, L.-C.; Van Speybroeck, V.; Head-Gordon, M.; Bell, A.T. Theoretical analysis of the influence of pore geometry on monomolecular cracking and dehydrogenation of n-butane in Brønsted acidic zeolites. ACS Catal. 2017, 7, 2685–2697.
- Janda, A.; Vlaisavljevich, B.; Lin, L.-C.; Mallikarjun Sharada, S.; Smit, B.; Head-Gordon, M.; Bell, A.T. Adsorption thermo-dynamics and intrinsic activation parameters for monomolecular cracking of n-alkanes on Brønsted acid sites in zeolites. J. Phys. Chem. C 2015, 119, 10427–10438.
- Sklenak, S.; Dedecek, J.; Li, C.; Wichterlova, B.; Gábová, V.; Sierka, M.; Sauer, J. Aluminium siting in the ZSM-5 framework by combination of high resolution 27Al NMR and DFT/MM calculations. Phys. Chem. Chem. Phys. 2009, 11, 1237–1247.
- Yang, C.-T.; Janda, A.; Bell, A.T.; Lin, L.-C. Atomistic investigations of the effects of Si/Al ratio and Al distribution on the adsorption selectivity of n-alkanes in Brønsted-acid zeolites. J. Phys. Chem. C 2018, 122, 9397–9410.
- Xu, B.; Sievers, C.; Hong, S.B.; Prins, R.; van Bokhoven, J.A. Catalytic activity of Brønsted acid sites in zeolites: Intrinsic activity, rate-limiting step, and influence of the local structure of the acid sites. J. Catal. 2006, 244, 163–168.
- Jones, A.J.; Zones, S.I.; Iglesia, E. Implications of transition state confinement within small voids for acid catalysis. J. Phys. Chem. C 2014, 118, 17787–17800.
- Van Bokhoven, J.A.; Xu, B. Towards predicting catalytic performances of zeolites. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 2007; Volume 170, pp. 1167–1173.
- Gounder, R.; Iglesia, E. Catalytic consequences of spatial constraints and acid site location for monomolecular alkane activation on zeolites. J. Am. Chem. Soc. 2009, 131, 1958–1971.
- Kadam, S.A.; Li, H.; Wormsbecher, R.F.; Travert, A. Impact of zeolite structure on entropic-enthalpic contributions to alkane monomolecular cracking: An IR operando study. Chem. Eur. J. 2018, 24, 5489–5492.
- Berger, F.; Rybicki, M.; Sauer, J. Adsorption and cracking of propane by zeolites of different pore size. J. Catal. 2021, 395, 117–128.
- Janda, A.; Bell, A. Effects of Si/Al ratio on the distribution of framework Al and on the rates of alkane monomolecular cracking and dehydrogenation in H-MFI. J. Am. Chem. Soc. 2013, 135, 19193–19207.
- Mallikarjun Sharada, S.; Zimmerman, P.M.; Bell, A.T.; Head-Gordon, M. Insights into the kinetics of cracking and dehydro-genation reactions of light alkanes in H-MFI. J. Phys. Chem. C 2013, 117, 12600–12611.
- First, E.L.; Gounaris, C.; Wei, J.; Floudas, C.A. Computational characterization of zeolite porous networks: An automated approach. Phys. Chem. Chem. Phys. 2011, 13, 17339–17358.
- Garcia-Martinez, J.; Johnson, M.M.; Valla, J.; Li, K.; Ying, J. Mesostructured zeolite Y—High hydrothermal stability and superior FCC catalytic performance. Catal. Sci. Technol. 2012, 2, 987–994.
- Choi, M.; Na, K.; Kim, J.; Sakamoto, Y.; Terasaki, O.; Ryoo, R. Stable single-unit-cell nanosheets of zeolite MFI as active and long-lived catalysts. Nature 2009, 461, 246–249.
- Li, K.; Valla, J.; Garcia-Martinez, J. Realizing the commercial potential of hierarchical zeolites: New opportunities in catalytic cracking. ChemCatChem 2013, 6, 46–66.
- Srivastava, R. Synthesis and applications of ordered and disordered mesoporous zeolites: Present and future prospective. Catal. Today 2018, 309, 172–188.
- Peng, P.; Gao, X.-H.; Yan, Z.-F.; Mintova, S. Diffusion and catalyst efficiency in hierarchical zeolite catalysts. Natl. Sci. Rev. 2020, 7, 1726–1742.
- Bai, P.; Haldoupis, E.; Dauenhauer, P.J.; Tsapatsis, M.; Siepmann, J.I. Understanding diffusion in hierarchical zeolites with house-of-cards nanosheets. ACS Nano 2016, 10, 7612–7618.
- Bu, L.; Nimlos, M.R.; Robichaud, D.J.; Kim, S. Diffusion of aromatic hydrocarbons in hierarchical mesoporous H-ZSM-5 zeolite. Catal. Today 2018, 312, 73–81.
- Grimme, S. Supramolecular binding thermodynamics by dispersion-corrected density functional theory. Chem. Eur. J. 2012, 18, 9955–9964.
- Josephson, T.R.; Dauenhauer, P.J.; Tsapatsis, M.; Siepmann, J.I. Adsorption of furan, hexanoic acid, and alkanes in a hierarchical zeolite at reaction conditions: Insights from molecular simulations. J. Comput. Sci. 2021, 48, 101267.
- Shetsiri, S.; Thivasasith, A.; Saenluang, K.; Wannapakdee, W.; Salakhum, S.; Wetchasat, P.; Nokbin, S.; Limtrakul, J.; Wattanakit, C. Sustainable production of ethylene from bioethanol over hierarchical ZSM-5 nanosheets. Sustain. Energy Fuels 2018, 3, 115–126.
- Yeh, J.Y.; Li, S.C.; Chen, C.H.; Wu, K.C.W.; Li, Y.P. Quantum mechanical calculations for biomass valorization over Metal-Organic Frameworks (MOFs). Chem. Asian J. 2021, 16, 1049–1056.
- Li, Y.-P.; Gomes, J.; Mallikarjun Sharada, S.; Bell, A.T.; Head-Gordon, M. Improved force-field parameters for QM/MM simulations of the energies of adsorption for molecules in zeolites and a free rotor correction to the rigid rotor harmonic oscillator model for adsorption enthalpies. J. Phys. Chem. C 2015, 119, 1840–1850.
- Li, Y.-P.; Head-Gordon, M.; Bell, A. Analysis of the reaction mechanism and catalytic activity of metal-substituted beta zeolite for the isomerization of glucose to fructose. ACS Catal. 2014, 4, 1537–1545.
- Li, Y.-P.; Head-Gordon, M.; Bell, A.T. Computational study of p-Xylene synthesis from ethylene and 2,5-dimethylfuran catalyzed by H-BEA. J. Phys. Chem. C 2014, 118, 22090–22095.
- Li, Y.-P.; Head-Gordon, M.; Bell, A.T. Theoretical study of 4-(hydroxymethyl) benzoic acid synthesis from ethylene and 5-(hydroxymethyl) furoic acid catalyzed by Sn-BEA. ACS Catal. 2016, 6, 5052–5061.

