 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Juan Ramon Peinado | + 2870 word(s) | 2870 | 2021-09-19 14:58:51 | | | |
| 2 | Conner Chen | Meta information modification | 2870 | 2021-10-08 08:27:07 | | |
Video Upload Options
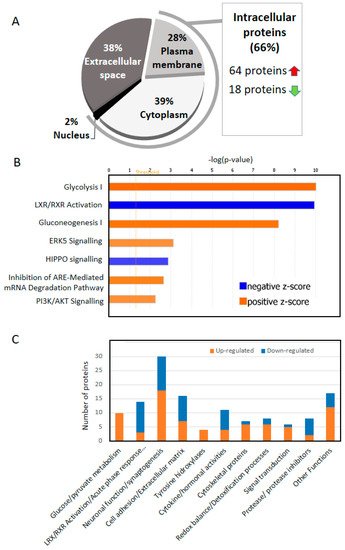
The fact that cerebrospinal fluid (CSF) deeply irrigates the brain together with the relative simplicity of sample extraction from patients make this biological fluid the best target for biomarker discovery in neurodegenerative diseases. Biomarker discovery has been especially fruitful for the identification new proteins that appear in the CSF of Alzheimer’s disease (AD) patients together with amyloid-β (Aβ42), total tau (T-tau), and phosphorylated tau (P-tau). Thus, several proteins have been already stablished as important biomarkers, due to an increase (i.e., CHI3L1) or a decrease (i.e., VGF) in AD patients’ CSF. Notwithstanding this, only a deep analysis of a database generated with all the changes observed in CSF across multiple proteomic studies, and especially those using state-of-the-art methodologies, may expose those components or metabolic pathways disrupted at different levels in AD. Deep comparative analysis of all the up- and down-regulated proteins across these studies revealed that 66% of the most consistent protein changes in CSF correspond to intracellular proteins.
1. Two Thirds of the of the Proteins That Change in the CSF of AD Are Intracellular
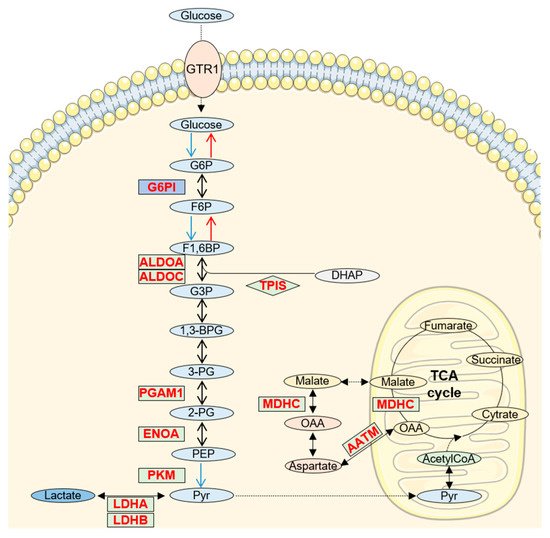
2. Increased Glucose/Pyruvate Metabolism in AD CSF
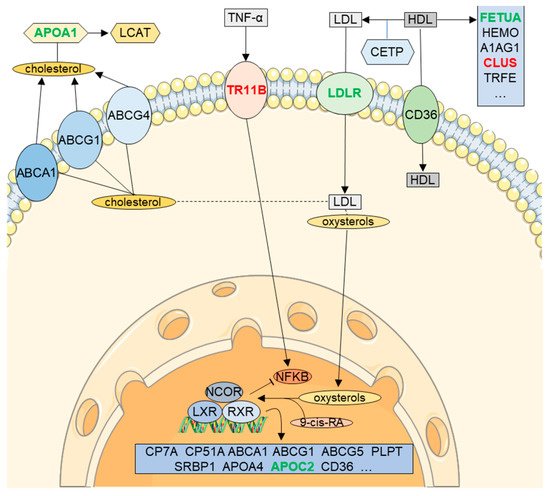
3. RXR Signaling in CSF (LXR/RXR Activation Pathway)
4. Neuronal Function/Synaptogenesis
5. 14-3-3 Proteins Are up Regulated in CSF
6. Cytokines and Hormones up and down Regulated in CSF
7. Importance of Cofactors in CSF
References
- Rahimi, J.; Woehrer, A. Overview of cerebrospinal fluid cytology. In Neuropathology; Kovacs, G., Alafuzoff, I., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 563–571.
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40.
- Reiber, H. Dynamics of brain-derived proteins in cerebrospinal fluid. Clin. Chim. Acta 2001, 310, 173–186.
- Stock, A.J.; Kasus-Jacobi, A.; Pereira, H.A. The role of neutrophil granule proteins in neuroinflammation and Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 240.
- Talib, L.L. Platelet biomarkers in Alzheimer’s disease. World J. Psychiatry 2012, 2, 95.
- Arai, K.; Gowert, N.S.; Donner, L.; Chatterjee, M.; Eisele, Y.S.; Towhid, S.T.; Münzer, P.; Walker, B.; Ogorek, I.; Borst, O.; et al. Blood Platelets in the Progression of Alzheimer’s Disease. PLoS ONE 2014, 9, e0090523.
- An, K.; Klyubin, I.; Kim, Y.; Jung, J.; Mably, A.J.; O’Dowd, S.T.; Lynch, T.; Kanmert, D.; Lemere, C.A.; Finan, G.M.; et al. Exosomes neutralize synaptic-plasticity-disrupting activity of Aβ assemblies in vivo. Mol. Brain 2013, 6, 47.
- Li, M.; Huang, L.; Chen, J.; Ni, F.; Zhang, Y.; Liu, F. Isolation of Exosome Nanoparticles from Human Cerebrospinal Fluid for Proteomic Analysis. ACS Appl. Nano Mater. 2021, 4, 3351–3359.
- Zahra, K.; Dey, T.; Ashish; Mishra, S.P.; Pandey, U. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front. Oncol. 2020, 10, 159.
- Bell, S.M.; Burgess, T.; Lee, J.; Blackburn, D.J.; Allen, S.P.; Mortiboys, H. Peripheral Glycolysis in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8924.
- Le Douce, J.; Maugard, M.; Veran, J.; Matos, M.; Jégo, P.; Vigneron, P.-A.; Faivre, E.; Toussay, X.; Vandenberghe, M.; Balbastre, Y.; et al. Impairment of Glycolysis-Derived l-Serine Production in Astrocytes Contributes to Cognitive Deficits in Alzheimer’s Disease. Cell Metab. 2020, 31, 503–517.e508.
- Bigl, M.; Bruckner, M.K.; Arendt, T.; Bigl, V.; Eschrich, K. Activities of key glycolytic enzymes in the brains of patients with Alzheimer’s disease. J. Neural. Transm. 1999, 106, 499–511.
- Goodman, A.B.; Pardee, A.B. Evidence for defective retinoid transport and function in late onset Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2901–2905.
- Ray, S.; Das, B.; Dasgupta, S. Potential therapeutic roles of retinoids for prevention of neuroinflammation and neurodegeneration in Alzheimer’s disease. Neural Regen. Res. 2019, 14, 1880.
- Kölsch, H.; Lütjohann, D.; Jessen, F.; Popp, J.; Hentschel, F.; Kelemen, P.; Friedrichs, S.; Maier, T.A.W.; Heun, R. RXRA gene variations influence Alzheimer’s disease risk and cholesterol metabolism. J. Cell. Mol. Med. 2009, 13, 589–598.
- Xiong, H.; Callaghan, D.; Jones, A.; Walker, D.G.; Lue, L.-F.; Beach, T.G.; Sue, L.I.; Woulfe, J.; Xu, H.; Stanimirovic, D.B.; et al. Cholesterol retention in Alzheimer’s brain is responsible for high β- and γ-secretase activities and Aβ production. Neurobiol. Dis. 2008, 29, 422–437.
- Ojo, J.O.; Crynen, G.; Reed, J.M.; Ajoy, R.; Vallabhaneni, P.; Algamal, M.; Leary, P.; Rafi, N.G.; Mouzon, B.; Mullan, M.; et al. Unbiased Proteomic Approach Identifies Unique and Coincidental Plasma Biomarkers in Repetitive mTBI and AD Pathogenesis. Front. Aging Neurosci. 2018, 10, 405.
- Sodhi, R.K.; Singh, N. Retinoids as potential targets for Alzheimer’s disease. Pharmacol. Biochem. Behav. 2014, 120, 117–123.
- Obulesu, M.; Dowlathabad, M.R.; Bramhachari, P.V. Carotenoids and Alzheimer’s Disease: An insight into therapeutic role of retinoids in animal models. Neurochem. Int. 2011, 59, 535–541.
- Fitz, N.F.; Nam, K.N.; Koldamova, R.; Lefterov, I. Therapeutic targeting of nuclear receptors, liver X and retinoid X receptors, for Alzheimer’s disease. Br. J. Pharmacol. 2019, 176, 3599–3610.
- Terwel, D.; Steffensen, K.R.; Verghese, P.B.; Kummer, M.P.; Gustafsson, J.A.; Holtzman, D.M.; Heneka, M.T. Critical Role of Astroglial Apolipoprotein E and Liver X Receptor- Expression for Microglial A Phagocytosis. J. Neurosci. 2011, 31, 7049–7059.
- Zelcer, N.; Khanlou, N.; Clare, R.; Jiang, Q.; Reed-Geaghan, E.G.; Landreth, G.E.; Vinters, H.V.; Tontonoz, P. Attenuation of neuroinflammation and Alzheimer’s disease pathology by liver x receptors. Proc. Natl. Acad. Sci. USA 2007, 104, 10601–10606.
- Andersson, S.; Gustafsson, N.; Warner, M.; Gustafsson, J.A. Inactivation of liver X receptor leads to adult-onset motor neuron degeneration in male mice. Proc. Natl. Acad. Sci. USA 2005, 102, 3857–3862.
- Zelcer, N. Liver X receptors as integrators of metabolic and inflammatory signaling. J. Clin. Investig. 2006, 116, 607–614.
- Dominiczak, M.H.; Caslake, M.J. Apolipoproteins: Metabolic role and clinical biochemistry applications. Ann. Clin. Biochem. Int. J. Lab. Med. 2011, 48, 498–515.
- Paula-Lima, A.C.; Tricerri, M.A.; Brito-Moreira, J.; Bomfim, T.R.; Oliveira, F.F.; Magdesian, M.H.; Grinberg, L.T.; Panizzutti, R.; Ferreira, S.T. Human apolipoprotein A–I binds amyloid-β and prevents Aβ-induced neurotoxicity. Int. J. Biochem. Cell Biol. 2009, 41, 1361–1370.
- Schellenberg, G.; Bird, T.; Wijsman, E.; Orr, H.; Anderson, L.; Nemens, E.; White, J.; Bonnycastle, L.; Weber, J.; Alonso, M.; et al. Genetic linkage evidence for a familial Alzheimer’s disease locus on chromosome 14. Science 1992, 258, 668–671.
- Friedman, D.J.; Pollak, M.R. APOL1and Kidney Disease: From Genetics to Biology. Annu. Rev. Physiol. 2020, 82, 323–342.
- Cohen-Cory, S. The developing synapse: Construction and modulation of synaptic structures and circuits. Science 2002, 298, 770–776.
- Masliah, E.; Mallory, M.; Alford, M.; DeTeresa, R.; Hansen, L.A.; McKeel, D.W.; Morris, J.C. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology 2001, 56, 127–129.
- Selkoe, D.J. Alzheimer’s Disease Is a Synaptic Failure. Science 2002, 298, 789–791.
- Pedrero-Prieto, C.M.; García-Carpintero, S.; Frontiñán-Rubio, J.; Llanos-González, E.; Aguilera García, C.; Alcaín, F.J.; Lindberg, I.; Durán-Prado, M.; Peinado, J.R.; Rabanal-Ruiz, Y. A comprehensive systematic review of CSF proteins and peptides that define Alzheimer’s disease. Clin. Proteom. 2020, 17, 1–24.
- Boles, N.C.; Hirsch, S.E.; Le, S.; Corneo, B.; Najm, F.; Minotti, A.P.; Wang, Q.; Lotz, S.; Tesar, P.J.; Fasano, C.A. NPTX1 Regulates Neural Lineage Specification from Human Pluripotent Stem Cells. Cell Rep. 2014, 6, 724–736.
- Sia, G.-M.; Béïque, J.-C.; Rumbaugh, G.; Cho, R.; Worley, P.F.; Huganir, R.L. Interaction of the N-Terminal Domain of the AMPA Receptor GluR4 Subunit with the Neuronal Pentraxin NP1 Mediates GluR4 Synaptic Recruitment. Neuron 2007, 55, 87–102.
- Cummings, D.M.; Benway, T.A.; Ho, H.; Tedoldi, A.; Fernandes Freitas, M.M.; Shahab, L.; Murray, C.E.; Richard-Loendt, A.; Brandner, S.; Lashley, T.; et al. Neuronal and Peripheral Pentraxins Modify Glutamate Release and may Interact in Blood–Brain Barrier Failure. Cereb. Cortex 2017, 27, 3437–3448.
- Lee, S.-J.; Wei, M.; Zhang, C.; Maxeiner, S.; Pak, C.; Calado Botelho, S.; Trotter, J.; Sterky, F.H.; Südhof, T.C. Presynaptic Neuronal Pentraxin Receptor Organizes Excitatory and Inhibitory Synapses. J. Neurosci. 2017, 37, 1062–1080.
- Wang, Z.; Wang, X.; Zou, H.; Dai, Z.; Feng, S.; Zhang, M.; Xiao, G.; Liu, Z.; Cheng, Q. The Basic Characteristics of the Pentraxin Family and Their Functions in Tumor Progression. Front. Immunol. 2020, 11, 1757.
- Yue, W.; Wang, T.; Zachariah, E.; Lin, Y.; Yang, C.S.; Xu, Q.; DiPaola, R.S.; Tan, X.-L. Transcriptomic analysis of pancreatic cancer cells in response to metformin and aspirin: An implication of synergy. Sci. Rep. 2015, 5, 1–11.
- Karagkounis, G.; Thai, L.; DeVecchio, J.; Gantt, G.A.; Duraes, L.; Pai, R.K.; Kalady, M.F. NPTX2 is associated with neoadjuvant therapy response in rectal cancer. J. Surg. Res. 2016, 202, 112–117.
- Lee, G.W.; Lee, T.H.; Vilcek, J. TSG-14, a tumor necrosis factor- and IL-1-inducible protein, is a novel member of the pentaxin family of acute phase proteins. J. Immunol. 1993, 150, 1804–1812.
- Xiao, M.-F.; Xu, D.; Craig, M.T.; Pelkey, K.A.; Chien, C.-C.; Shi, Y.; Zhang, J.; Resnick, S.; Pletnikova, O.; Salmon, D.; et al. NPTX2 and cognitive dysfunction in Alzheimer’s Disease. eLife 2017, 6, e23798.
- De Groot, J.; Sontheimer, H. Glutamate and the biology of gliomas. Glia 2011, 59, 1181–1189.
- Radin, D.P.; Patel, P. A current perspective on the oncopreventive and oncolytic properties of selective serotonin reuptake inhibitors. Biomed. Pharmacother. 2017, 87, 636–639.
- Abad, M.A.; Enguita, M.; DeGregorio-Rocasolano, N.; Ferrer, I.; Trullas, R. Neuronal Pentraxin 1 Contributes to the Neuronal Damage Evoked by Amyloid- and Is Overexpressed in Dystrophic Neurites in Alzheimer’s Brain. J. Neurosci. 2006, 26, 12735–12747.
- Brito-Moreira, J.; Lourenco, M.V.; Oliveira, M.M.; Ribeiro, F.C.; Ledo, J.H.; Diniz, L.P.; Vital, J.F.S.; Magdesian, M.H.; Melo, H.M.; Barros-Aragão, F.; et al. Interaction of amyloid-β (Aβ) oligomers with neurexin 2α and neuroligin 1 mediates synapse damage and memory loss in mice. J. Biol. Chem. 2017, 292, 7327–7337.
- Pettem, K.L.; Yokomaku, D.; Luo, L.; Linhoff, M.W.; Prasad, T.; Connor, S.A.; Siddiqui, T.J.; Kawabe, H.; Chen, F.; Zhang, L.; et al. The Specific α-Neurexin Interactor Calsyntenin-3 Promotes Excitatory and Inhibitory Synapse Development. Neuron 2013, 80, 113–128.
- Um, J.W.; Pramanik, G.; Ko, J.S.; Song, M.Y.; Lee, D.; Kim, H.; Park, K.S.; Südhof, T.C.; Tabuchi, K.; Ko, J. Calsyntenins Function as Synaptogenic Adhesion Molecules in Concert with Neurexins. Cell Rep. 2014, 6, 1096–1109.
- Hintsch, G. The Calsyntenins—A Family of Postsynaptic Membrane Proteins with Distinct Neuronal Expression Patterns. Mol. Cell. Neurosci. 2002, 21, 393–409.
- Uchida, Y.; Gomi, F. The role of calsyntenin-3 in dystrophic neurite formation in Alzheimer’s disease brain. Geriatr. Gerontol. Int. 2016, 16, 43–50.
- Pilozzi, A.; Carro, C.; Whalen, M.; Huang, X. Blood-Brain Barrier Degradation and the Implication of SPARC Protein as a Potential Therapeutic Target for Alzheimer’s Disease. In Alzheimer’s Disease: Drug Discovery; Huang, X., Ed.; Exon Publications: Brisbane, Australia, 2020.
- McCurdy, S.; Baicu, C.F.; Heymans, S.; Bradshaw, A.D. Cardiac extracellular matrix remodeling: Fibrillar collagens and Secreted Protein Acidic and Rich in Cysteine (SPARC). J. Mol. Cell. Cardiol. 2010, 48, 544–549.
- Giannoni, P.; Arango-Lievano, M.; Neves, I.D.; Rousset, M.-C.; Baranger, K.; Rivera, S.; Jeanneteau, F.; Claeysen, S.; Marchi, N. Cerebrovascular pathology during the progression of experimental Alzheimer’s disease. Neurobiol. Dis. 2016, 88, 107–117.
- Awwad, K.; Hu, J.; Shi, L.; Mangels, N.; Abdel Malik, R.; Zippel, N.; Fisslthaler, B.; Eble, J.A.; Pfeilschifter, J.; Popp, R.; et al. Role of secreted modular calcium-binding protein 1 (SMOC1) in transforming growth factor β signalling and angiogenesis. Cardiovasc. Res. 2015, 106, 284–294.
- Bai, B.; Wang, X.; Li, Y.; Chen, P.-C.; Yu, K.; Dey, K.K.; Yarbro, J.M.; Han, X.; Lutz, B.M.; Rao, S.; et al. Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer’s Disease Progression. Neuron 2020, 105, 975–991.e977.
- Sathe, G.; Albert, M.; Darrow, J.; Saito, A.; Troncoso, J.; Pandey, A.; Moghekar, A. Quantitative proteomic analysis of the frontal cortex in Alzheimer’s disease. J. Neurochem. 2020, 156, 988–1002.
- Barrera-Ocampo, A.; Arlt, S.; Matschke, J.; Hartmann, U.; Puig, B.; Ferrer, I.; Zürbig, P.; Glatzel, M.; Sepulveda-Falla, D.; Jahn, H. Amyloid-β Precursor Protein Modulates the Sorting of Testican-1 and Contributes to Its Accumulation in Brain Tissue and Cerebrospinal Fluid from Patients with Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2016, 75, 903–916.
- Schechter, I.; Ziv, E. Cathepsins S, B and L with aminopeptidases display beta-secretase activity associated with the pathogenesis of Alzheimer’s disease. Biol. Chem. 2011, 392, 555–569.
- Merlo, S.; Sortino, M.A. Estrogen activates matrix metalloproteinases-2 and -9 to increase beta amyloid degradation. Mol. Cell. Neurosci. 2012, 49, 423–429.
- Ichimura, T.; Isobe, T.; Okuyama, T.; Yamauchi, T.; Fujisawa, H. Brain 14-3-3 protein is an activator protein that activates tryptophan 5-monooxygenase and tyrosine 3-monooxygenase in the presence of Ca2+,calmodulin-dependent protein kinase II. FEBS Lett. 1987, 219, 79–82.
- Cho, E.; Park, J.-Y. Emerging roles of 14-3-3γ in the brain disorder. BMB Rep. 2020, 53, 500–511.
- Cornell, B.; Toyo-oka, K. 14-3-3 Proteins in Brain Development: Neurogenesis, Neuronal Migration and Neuromorphogenesis. Front. Mol. Neurosci. 2017, 10, 318.
- Foote, M.; Zhou, Y. 14-3-3 proteins in neurological disorders. Int. J. Biochem. Mol. Biol. 2012, 3, 152–164.
- Kelly, J.; Moyeed, R.; Carroll, C.; Albani, D.; Li, X. Gene expression meta-analysis of Parkinson’s disease and its relationship with Alzheimer’s disease. Mol. Brain 2019, 12, 1–10.
- Bartolomucci, A.; Pasinetti, G.M.; Salton, S.R.J. Granins as disease-biomarkers: Translational potential for psychiatric and neurological disorders. Neuroscience 2010, 170, 289–297.
- Bartolomucci, A.; Possenti, R.; Mahata, S.K.; Fischer-Colbrie, R.; Loh, Y.P.; Salton, S.R.J. The Extended Granin Family: Structure, Function, and Biomedical Implications. Endocr. Rev. 2011, 32, 755–797.
- Serby, M.; Richardson, S.B.; Twente, S.; Siekierski, J.; Corwin, J.; Rotrosen, J. CSF somatostatin in Alzheimer’s disease. Neurobiol. Aging 1984, 5, 187–189.
- Sagar, S.M.; Flint Beal, M.; Marshall, P.E.; Landis, D.M.D.; Martin, J.B. Implications of neuropeptides in neurological diseases. Peptides 1984, 5, 255–262.
- Paik, S.; Somvanshi, R.K.; Kumar, U. Somatostatin-Mediated Changes in Microtubule-Associated Proteins and Retinoic Acid–Induced Neurite Outgrowth in SH-SY5Y Cells. J. Mol. Neurosci. 2019, 68, 120–134.
- Sodhi, C.P.; Phadke, S.A.; Batlle, D.; Sahai, A. Hypoxia Stimulates Osteopontin Expression and Proliferation of Cultured Vascular Smooth Muscle Cells. Diabetes 2001, 50, 1482–1490.
- Rabenstein, M.; Vay, S.U.; Flitsch, L.J.; Fink, G.R.; Schroeter, M.; Rueger, M.A. Osteopontin directly modulates cytokine expression of primary microglia and increases their survival. J. Neuroimmunol. 2016, 299, 130–138.
- Wung, J.; Perry, G.; Kowalski, A.; Harris, P.R.; Bishop, G.; Trivedi, M.; Johnson, S.; Smith, M.; Denhardt, D.; Atwood, C. Increased Expression of the Remodeling- and Tumorigenic-Associated Factor Osteopontin in Pyramidal Neurons of the Alzheimers Disease Brain. Curr. Alzheimer Res. 2007, 4, 67–72.
- Comi, C.; Carecchio, M.; Chiocchetti, A.; Nicola, S.; Galimberti, D.; Fenoglio, C.; Cappellano, G.; Monaco, F.; Scarpini, E.; Dianzani, U. Osteopontin is Increased in the Cerebrospinal Fluid of Patients with Alzheimer’s Disease and Its Levels Correlate with Cognitive Decline. J. Alzheimer’s Dis. 2010, 19, 1143–1148.
- Sensi, S.L.; Granzotto, A.; Siotto, M.; Squitti, R. Copper and Zinc Dysregulation in Alzheimer’s Disease. Trends. Pharmacol. Sci. 2018, 39, 1049–1063.
- Mold, M.; Ouro-Gnao, L.; Wieckowski, B.M.; Exley, C. Copper prevents amyloid-β1–42 from forming amyloid fibrils under near-physiological conditions in vitro. Sci. Rep. 2013, 3, 1256.
- Singh, I.; Sagare, A.P.; Coma, M.; Perlmutter, D.; Gelein, R.; Bell, R.D.; Deane, R.J.; Zhong, E.; Parisi, M.; Ciszewski, J.; et al. Low levels of copper disrupt brain amyloid-β homeostasis by altering its production and clearance. Proc. Natl. Acad. Sci. USA 2013, 110, 14771–14776.
- Bagheri, S.; Squitti, R.; Haertlé, T.; Siotto, M.; Saboury, A.A. Role of Copper in the Onset of Alzheimer’s Disease Compared to Other Metals. Front. Aging Neurosci. 2018, 9, 446.
- Roberts, B.R.; Ryan, T.M.; Bush, A.I.; Masters, C.L.; Duce, J.A. The role of metallobiology and amyloid-beta peptides in Alzheimer’s disease. J. Neurochem. 2012, 120 (Suppl. 1), 149–166.
- Twomey, P.J.; Viljoen, A.; House, I.M.; Reynolds, T.M.; Wierzbicki, A.S. Relationship between serum copper, ceruloplasmin, and non-ceruloplasmin-bound copper in routine clinical practice. Clin. Chem. 2005, 51, 1558–1559.
- Vašák, M.; Meloni, G. Mammalian Metallothionein-3: New Functional and Structural Insights. Int. J. Mol. Sci. 2017, 18, 1117.
- Eipper, B.A.; Mains, R.E. Peptide α-Amidation. Annu. Rev. Physiol. 1988, 50, 333–344.
- Eipper, B.A.; Stoffers, D.A.; Mains, R.E. The biosynthesis of neuropeptides: Peptide alpha-amidation. Annu. Rev. Neurosci. 1992, 15, 57–85.
- Wand, G.S.; May, C.; May, V.; Whitehouse, P.J.; Rapoport, S.I.; Eipper, B.A. Alzheimer’s disease: Low levels of peptide alpha-amidation activity in brain and CSF. Neurology 1987, 37, 1057–1061.


