 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Monica Neagu | + 7829 word(s) | 7829 | 2021-09-19 15:27:57 | | | |
| 2 | Conner Chen | Meta information modification | 7829 | 2021-10-08 08:05:23 | | |
Video Upload Options
Cutaneous melanoma (CM) is an aggressive neoplasm that evolves from the malignant transformation of neural crest stem cell-derived melanocytes. The etiology of melanoma is multifactorial, and the most prominent factors include genetic predisposition, light skin color, multiple naevi, and excessive exposure to UV. Epigenetic alterations have emerged as essential contributors in the pathogenesis of various human diseases, including CM. Epigenetics is another layer of instructions apart from the genetic code that controls how genes are read and expressed, involving a change in the cell phenotype without changes in the genotype. Epigenetic regulation is an umbrella term that encompasses several mechanisms such as DNA methylation, histone post-translational modifications (PTMs), nucleosome remodeling, histone variants, and RNA-mediated post-transcriptional regulation. Influenced by lifestyle and environmental factors, epigenetic changes are highly dynamic and reversible and thus easy to regulate.
1. CM Epigenetics
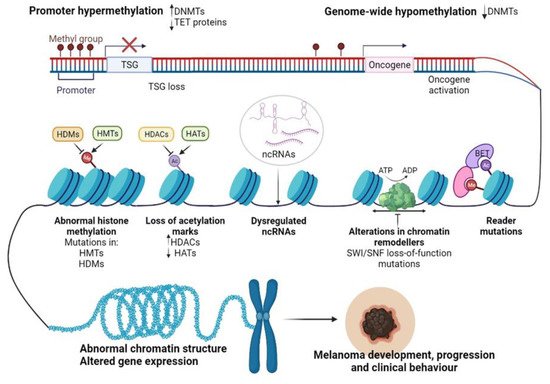
2. Epigenetic Alterations Driving CM initiation and Progression
2.1. DNA Methylation in CM Development
2.2. Histone-Modifying Enzymes and PTMs in CM Development
2.2.1. Histone Modifications “Writers”
H3K4 Methyltransferases (KMT2D)
The Roles of H3K4 Methylation Marks
H3K27 Methyltransferases (EZH2)
H3K9 Methyltransferases
2.2.2. Histone Modifications “Readers”
2.2.3. Histone Modifications “Erasers”
Histone Deacetylases (HDACs)
Histone Demethylases (HDMs)
3. Epigenetic Alterations Involved in CM Drug Resistance
3.1. Resistance to MAPK Inhibitors (MAPKi)
3.1.1. DNA Methylation and MAPKi Resistance
3.1.2. Histone-Modifying Enzymes and PTMs Involved in MAPKi Resistance
3.2. Resistance to Immunotherapy
3.2.1. DNA Methylation and Resistance to Immunotherapy
3.2.2. Histone-Modifying Enzymes and PTMs Involved in Immunotherapy Resistance
References
- Farooqi, A.A.; Fayyaz, S.; Poltronieri, P.; Calin, G.; Mallardo, M. Epigenetic deregulation in cancer: Enzyme players and non-coding RNAs. Semin. Cancer Biol. 2020.
- de Unamuno Bustos, B.; Murria Estal, R.; Pérez Simó, G.; Simarro Farinos, J.; Pujol Marco, C.; Navarro Mira, M.; Alegre de Miquel, V.; Ballester Sánchez, R.; Sabater Marco, V.; Llavador Ros, M.; et al. Aberrant DNA methylation is associated with aggressive clinicopathological features and poor survival in cutaneous melanoma. Br. J. Dermatol. 2018, 179, 394–404.
- Kampilafkos, P.; Melachrinou, M.; Kefalopoulou, Z.; Lakoumentas, J.; Sotiropoulou-Bonikou, G. Epigenetic modifications in cutaneous malignant melanoma: EZH2, H3K4me2, and H3K27me3 immunohistochemical expression is enhanced at the invasion front of the tumor. Am. J. Dermatopathol. 2015, 37, 138–144.
- Maitituoheti, M.; Keung, E.Z.; Tang, M.; Yan, L.; Alam, H.; Han, G.; Singh, A.K.; Raman, A.T.; Terranova, C.; Sarkar, S.; et al. Enhancer Reprogramming Confers Dependence on Glycolysis and IGF Signaling in KMT2D Mutant Melanoma. Cell Rep. 2020, 33, 108293.
- Neagu, M.; Constantin, C.; Cretoiu, S.M.; Zurac, S. miRNAs in the Diagnosis and Prognosis of Skin Cancer. Front. Cell Dev. Biol. 2020, 8, 71.
- Yu, Y.; Schleich, K.; Yue, B.; Ji, S.; Lohneis, P.; Kemper, K.; Silvis, M.R.; Qutob, N.; van Rooijen, E.; Werner-Klein, M.; et al. Targeting the Senescence-Overriding Cooperative Activity of Structurally Unrelated H3K9 Demethylases in Melanoma. Cancer Cell 2018, 33, 322–336.e8.
- Zingg, D.; Debbache, J.; Schaefer, S.M.; Tuncer, E.; Frommel, S.C.; Cheng, P.; Arenas-Ramirez, N.; Haeusel, J.; Zhang, Y.; Bonalli, M.; et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat. Commun. 2015, 6, 6051.
- Zingg, D.; Debbache, J.; Peña-Hernández, R.; Antunes, A.T.; Schaefer, S.M.; Cheng, P.F.; Zimmerli, D.; Haeusel, J.; Calçada, R.R.; Tuncer, E.; et al. EZH2-Mediated Primary Cilium Deconstruction Drives Metastatic Melanoma Formation. Cancer Cell 2018, 34, 69–84.e14.
- Liu, X.; Zhang, S.-M.; McGeary, M.K.; Krykbaeva, I.; Lai, L.; Jansen, D.J.; Kales, S.C.; Simeonov, A.; Hall, M.D.; Kelly, D.P.; et al. KDM5B Promotes Drug Resistance by Regulating Melanoma-Propagating Cell Subpopulations. Mol. Cancer Ther. 2019, 18, 706–717.
- Licht, J.D.; Bennett, R.L. Leveraging epigenetics to enhance the efficacy of immunotherapy. Clin. Epigenetics 2021, 13, 115.
- Maiuri, A.R.; O’Hagan, H.M. Interplay Between Inflammation and Epigenetic Changes in Cancer. Prog. Mol. Biol. Transl. Sci. 2016, 144, 69–117.
- Maes, K.; Mondino, A.; Lasarte, J.J.; Agirre, X.; Vanderkerken, K.; Prosper, F.; Breckpot, K. Epigenetic Modifiers: Anti-Neoplastic Drugs With Immunomodulating Potential. Front. Immunol. 2021, 12, 652160.
- Kominsky, D.J.; Campbell, E.L.; Colgan, S.P. Metabolic shifts in immunity and inflammation. J. Immunol. 2010, 184, 4062–4068.
- Johnson, C.; Warmoes, M.O.; Shen, X.; Locasale, J.W. Epigenetics and cancer metabolism. Cancer Lett. 2015, 356, 309–314.
- Franco, R.; Schoneveld, O.; Georgakilas, A.G.; Panayiotidis, M.I. Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett. 2008, 266, 6–11.
- Ciążyńska, M.; Olejniczak-Staruch, I.; Sobolewska-Sztychny, D.; Narbutt, J.; Skibińska, M.; Lesiak, A. Ultraviolet Radiation and Chronic Inflammation-Molecules and Mechanisms Involved in Skin Carcinogenesis: A Narrative Review. Life 2021, 11, 326.
- Fan, Y.; Mao, R.; Yang, J. NF-κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell 2013, 4, 176–185.
- Micevic, G.; Theodosakis, N.; Bosenberg, M. Aberrant DNA methylation in melanoma: Biomarker and therapeutic opportunities. Clin. Epigenetics 2017, 9, 34.
- Howell, P.M.J.; Liu, S.; Ren, S.; Behlen, C.; Fodstad, O.; Riker, A.I. Epigenetics in human melanoma. Cancer Control 2009, 16, 200–218.
- Hoon, D.S.B.; Spugnardi, M.; Kuo, C.; Huang, S.K.; Morton, D.L.; Taback, B. Profiling epigenetic inactivation of tumor suppressor genes in tumors and plasma from cutaneous melanoma patients. Oncogene 2004, 23, 4014–4022.
- Palanca-Ballester, C.; Rodriguez-Casanova, A.; Torres, S.; Calabuig-Fariñas, S.; Exposito, F.; Serrano, D.; Redin, E.; Valencia, K.; Jantus-Lewintre, E.; Diaz-Lagares, A.; et al. Cancer Epigenetic Biomarkers in Liquid Biopsy for High Incidence Malignancies. Cancers 2021, 13, 3016.
- Tanemura, A.; Terando, A.M.; Sim, M.-S.; van Hoesel, A.Q.; de Maat, M.F.G.; Morton, D.L.; Hoon, D.S.B. CpG island methylator phenotype predicts progression of malignant melanoma. Clin. cancer Res. an Off. J. Am. Assoc. Cancer Res. 2009, 15, 1801–1807.
- Garcia-Alvarez, A.; Ortiz, C.; Muñoz-Couselo, E. Current Perspectives and Novel Strategies of NRAS-Mutant Melanoma. Onco. Targets. Ther. 2021, 14, 3709–3719.
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259.
- Ponomaryova, A.A.; Rykova, E.Y.; Gervas, P.A.; Cherdyntseva, N.V.; Mamedov, I.Z.; Azhikina, T.L. Aberrant Methylation of LINE-1 Transposable Elements: A Search for Cancer Biomarkers. Cells 2020, 9, 2017.
- Ecsedi, S.I.; Hernandez-Vargas, H.; Lima, S.C.; Herceg, Z.; Adany, R.; Balazs, M. Transposable hypomethylation is associated with metastatic capacity of primary melanomas. Int. J. Clin. Exp. Pathol. 2013, 6, 2943–2948.
- De Araújo, É.S.S.; Kashiwabara, A.Y.; Achatz, M.I.W.; Moredo, L.F.; De Sá, B.C.S.; Duprat, J.P.; Rosenberg, C.; Carraro, D.M.; Krepischi, A.C. V LINE-1 hypermethylation in peripheral blood of cutaneous melanoma patients is associated with metastasis. Melanoma Res. 2015, 25, 173–177.
- Sigalotti, L.; Fratta, E.; Bidoli, E.; Covre, A.; Parisi, G.; Colizzi, F.; Coral, S.; Massarut, S.; Kirkwood, J.M.; Maio, M. Methylation levels of the “long interspersed nucleotide element-1” repetitive sequences predict survival of melanoma patients. J. Transl. Med. 2011, 9, 78.
- Tonella, L.; Pala, V.; Ponti, R.; Rubatto, M.; Gallo, G.; Mastorino, L.; Avallone, G.; Merli, M.; Agostini, A.; Fava, P.; et al. Prognostic and Predictive Biomarkers in Stage III Melanoma: Current Insights and Clinical Implications. Int. J. Mol. Sci. 2021, 22, 4561.
- Colemon, A.; Harris, T.M.; Ramanathan, S. DNA hypomethylation drives changes in MAGE-A gene expression resulting in alteration of proliferative status of cells. Genes Environ. Off. J. Japanese Environ. Mutagen Soc. 2020, 42, 24.
- Faramarzi, S.; Ghafouri-Fard, S. Melanoma: A prototype of cancer-testis antigen-expressing malignancies. Immunotherapy 2017, 9, 1103–1113.
- Sigalotti, L.; Coral, S.; Nardi, G.; Spessotto, A.; Cortini, E.; Cattarossi, I.; Colizzi, F.; Altomonte, M.; Maio, M. Promoter methylation controls the expression of MAGE2, 3 and 4 genes in human cutaneous melanoma. J. Immunother. 2002, 25, 16–26.
- Sigalotti, L.; Covre, A.; Zabierowski, S.; Himes, B.; Colizzi, F.; Natali, P.G.; Herlyn, M.; Maio, M. Cancer testis antigens in human melanoma stem cells: Expression, distribution, and methylation status. J. Cell. Physiol. 2008, 215, 287–291.
- Danilova, A.; Misyurin, V.; Novik, A.; Girdyuk, D.; Avdonkina, N.; Nekhaeva, T.; Emelyanova, N.; Pipia, N.; Misyurin, A.; Baldueva, I. Cancer/testis antigens expression during cultivation of melanoma and soft tissue sarcoma cells. Clin. Sarcoma Res. 2020, 10, 3.
- Fujiwara, S.; Nagai, H.; Jimbo, H.; Jimbo, N.; Tanaka, T.; Inoie, M.; Nishigori, C. Gene Expression and Methylation Analysis in Melanomas and Melanocytes From the Same Patient: Loss of NPM2 Expression Is a Potential Immunohistochemical Marker for Melanoma. Front. Oncol. 2018, 8, 675.
- Gao, L.; van den Hurk, K.; Moerkerk, P.T.M.; Goeman, J.J.; Beck, S.; Gruis, N.A.; van den Oord, J.J.; Winnepenninckx, V.J.; van Engeland, M.; van Doorn, R. Promoter CpG island hypermethylation in dysplastic nevus and melanoma: CLDN11 as an epigenetic biomarker for malignancy. J. Invest. Dermatol. 2014, 134, 2957–2966.
- Lauss, M.; Ringnér, M.; Karlsson, A.; Harbst, K.; Busch, C.; Geisler, J.; Lønning, P.E.; Staaf, J.; Jönsson, G. DNA methylation subgroups in melanoma are associated with proliferative and immunological processes. BMC Med. Genom. 2015, 8, 73.
- Yamamoto, Y.; Matsusaka, K.; Fukuyo, M.; Rahmutulla, B.; Matsue, H.; Kaneda, A. Higher methylation subtype of malignant melanoma and its correlation with thicker progression and worse prognosis. Cancer Med. 2020, 9, 7194–7204.
- Marzese, D.M.; Scolyer, R.A.; Huynh, J.L.; Huang, S.K.; Hirose, H.; Chong, K.K.; Kiyohara, E.; Wang, J.; Kawas, N.P.; Donovan, N.C.; et al. Epigenome-wide DNA methylation landscape of melanoma progression to brain metastasis reveals aberrations on homeobox D cluster associated with prognosis. Hum. Mol. Genet. 2014, 23, 226–238.
- Lian, C.G.; Xu, Y.; Ceol, C.; Wu, F.; Larson, A.; Dresser, K.; Xu, W.; Tan, L.; Hu, Y.; Zhan, Q.; et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 2012, 150, 1135–1146.
- Saldanha, G.; Joshi, K.; Lawes, K.; Bamford, M.; Moosa, F.; Teo, K.W.; Pringle, J.H. 5-Hydroxymethylcytosine is an independent predictor of survival in malignant melanoma. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc 2017, 30, 60–68.
- Khaliq, M.; Fallahi-Sichani, M. Epigenetic Mechanisms of Escape from BRAF Oncogene Dependency. Cancers 2019, 11, 1480.
- Strub, T.; Ballotti, R.; Bertolotto, C. The “ART” of Epigenetics in Melanoma: From histone “Alterations, to Resistance and Therapies”. Theranostics 2020, 10, 1777–1797.
- Frantz, W.T.; Ceol, C.J. From Tank to Treatment: Modeling Melanoma in Zebrafish. Cells 2020, 9, 1289.
- Patton, E.E.; Widlund, H.R.; Kutok, J.L.; Kopani, K.R.; Amatruda, J.F.; Murphey, R.D.; Berghmans, S.; Mayhall, E.A.; Traver, D.; Fletcher, C.D.M.; et al. BRAF mutations are sufficient to promote nevi formation and cooperate with p53 in the genesis of melanoma. Curr. Biol. 2005, 15, 249–254.
- Scahill, C.M.; Digby, Z.; Sealy, I.M.; Wojciechowska, S.; White, R.J.; Collins, J.E.; Stemple, D.L.; Bartke, T.; Mathers, M.E.; Patton, E.E.; et al. Loss of the chromatin modifier Kdm2aa causes BrafV600E-independent spontaneous melanoma in zebrafish. PLoS Genet. 2017, 13, e1006959.
- Herz, H.-M.; Mohan, M.; Garruss, A.S.; Liang, K.; Takahashi, Y.-H.; Mickey, K.; Voets, O.; Verrijzer, C.P.; Shilatifard, A. Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev. 2012, 26, 2604–2620.
- Wang, C.; Lee, J.-E.; Lai, B.; Macfarlan, T.S.; Xu, S.; Zhuang, L.; Liu, C.; Peng, W.; Ge, K. Enhancer priming by H3K4 methyltransferase MLL4 controls cell fate transition. Proc. Natl. Acad. Sci. USA 2016, 113, 11871–11876.
- Cheng, C.S.; Rai, K.; Garber, M.; Hollinger, A.; Robbins, D.; Anderson, S.; Macbeth, A.; Tzou, A.; Carneiro, M.O.; Raychowdhury, R.; et al. Semiconductor-based DNA sequencing of histone modification states. Nat. Commun. 2013, 4, 2672.
- Bossi, D.; Cicalese, A.; Dellino, G.I.; Luzi, L.; Riva, L.; D’Alesio, C.; Diaferia, G.R.; Carugo, A.; Cavallaro, E.; Piccioni, R.; et al. In Vivo Genetic Screens of Patient-Derived Tumors Revealed Unexpected Frailty of the Transformed Phenotype. Cancer Discov. 2016, 6, 650–663.
- Gu, B.; Lee, M.G. Histone H3 lysine 4 methyltransferases and demethylases in self-renewal and differentiation of stem cells. Cell Biosci. 2013, 3, 39.
- Bernstein, B.E.; Kamal, M.; Lindblad-Toh, K.; Bekiranov, S.; Bailey, D.K.; Huebert, D.J.; McMahon, S.; Karlsson, E.K.; Kulbokas, E.J., 3rd; Gingeras, T.R.; et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 2005, 120, 169–181.
- Uzdensky, A.; Demyanenko, S.; Bibov, M.; Sharifulina, S.; Kit, O.; Przhedetski, Y.; Pozdnyakova, V. Expression of proteins involved in epigenetic regulation in human cutaneous melanoma and peritumoral skin. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2014, 35, 8225–8233.
- Terranova, C.; Tang, M.; Maitituoheti, M.; Raman, A.T.; Schulz, J.; Amin, S.B.; Orouji, E.; Tomczak, K.; Sarkar, S.; Oba, J.; et al. Bivalent and broad chromatin domains regulate pro-metastatic drivers in melanoma. bioRxiv 2019, 721480.
- Neagu, M.; Constantin, C.; Bostan, M.; Caruntu, C.; Ignat, S.R.; Dinescu, S.; Costache, M. Proteomic Technology “Lens” for Epithelial-Mesenchymal Transition Process Identification in Oncology. Anal. Cell. Pathol. 2019, 2019, 3565970.
- Hoffmann, F.; Niebel, D.; Aymans, P.; Ferring-Schmitt, S.; Dietrich, D.; Landsberg, J. H3K27me3 and EZH2 expression in melanoma: Relevance for melanoma progression and response to immune checkpoint blockade. Clin. Epigenetics 2020, 12, 24.
- McHugh, J.B.; Fullen, D.R.; Ma, L.; Kleer, C.G.; Su, L.D. Expression of polycomb group protein EZH2 in nevi and melanoma. J. Cutan. Pathol. 2007, 34, 597–600.
- Bachmann, I.M.; Halvorsen, O.J.; Collett, K.; Stefansson, I.M.; Straume, O.; Haukaas, S.A.; Salvesen, H.B.; Otte, A.P.; Akslen, L.A. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 268–273.
- Fan, T.; Jiang, S.; Chung, N.; Alikhan, A.; Ni, C.; Lee, C.-C.R.; Hornyak, T.J. EZH2-dependent suppression of a cellular senescence phenotype in melanoma cells by inhibition of p21/CDKN1A expression. Mol. Cancer Res. 2011, 9, 418–429.
- De Donatis, G.M.; Le Pape, E.; Pierron, A.; Cheli, Y.; Hofman, V.; Hofman, P.; Allegra, M.; Zahaf, K.; Bahadoran, P.; Rocchi, S.; et al. NF-kB2 induces senescence bypass in melanoma via a direct transcriptional activation of EZH2. Oncogene 2016, 35, 2735–2745.
- Barsotti, A.M.; Ryskin, M.; Zhong, W.; Zhang, W.-G.; Giannakou, A.; Loreth, C.; Diesl, V.; Follettie, M.; Golas, J.; Lee, M.; et al. Epigenetic reprogramming by tumor-derived EZH2 gain-of-function mutations promotes aggressive 3D cell morphologies and enhances melanoma tumor growth. Oncotarget 2015, 6, 2928–2938.
- Ceol, C.J.; Houvras, Y.; Jane-Valbuena, J.; Bilodeau, S.; Orlando, D.A.; Battisti, V.; Fritsch, L.; Lin, W.M.; Hollmann, T.J.; Ferré, F.; et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature 2011, 471, 513–517.
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J. 3rd SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932.
- Orouji, E.; Utikal, J. Tackling malignant melanoma epigenetically: Histone lysine methylation. Clin. Epigenetics 2018, 10, 145.
- Kostaki, M.; Manona, A.D.; Stavraka, I.; Korkolopoulou, P.; Levidou, G.; Trigka, E.-A.; Christofidou, E.; Champsas, G.; Stratigos, A.J.; Katsambas, A.; et al. High-frequency p16(INK) (4A) promoter methylation is associated with histone methyltransferase SETDB1 expression in sporadic cutaneous melanoma. Exp. Dermatol. 2014, 23, 332–338.
- Orouji, E.; Federico, A.; Larribère, L.; Novak, D.; Lipka, D.B.; Assenov, Y.; Sachindra, S.; Hüser, L.; Granados, K.; Gebhardt, C.; et al. Histone methyltransferase SETDB1 contributes to melanoma tumorigenesis and serves as a new potential therapeutic target. Int. J. Cancer 2019, 145, 3462–3477.
- Gillette, T.G.; Hill, J.A. Readers, writers, and erasers: Chromatin as the whiteboard of heart disease. Circ. Res. 2015, 116, 1245–1253.
- Trivedi, A.; Mehrotra, A.; Baum, C.E.; Lewis, B.; Basuroy, T.; Blomquist, T.; Trumbly, R.; Filipp, F.V.; Setaluri, V.; De La Serna, I.L. Bromodomain and extra-terminal domain (BET) proteins regulate melanocyte differentiation. Epigenetics Chromatin 2020, 13, 1–18.
- Denis, G.V.; McComb, M.E.; Faller, D.V.; Sinha, A.; Romesser, P.B.; Costello, C.E. Identification of transcription complexes that contain the double bromodomain protein Brd2 and chromatin remodeling machines. J. Proteome Res. 2006, 5, 502–511.
- Segura, M.F.; Fontanals-Cirera, B.; Gaziel-Sovran, A.; Guijarro, M.V.; Hanniford, D.; Zhang, G.; González-Gomez, P.; Morante, M.; Jubierre, L.; Zhang, W.; et al. BRD4 sustains melanoma proliferation and represents a new target for epigenetic therapy. Cancer Res. 2013, 73, 6264–6276.
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6.
- Fiziev, P.; Akdemir, K.C.; Miller, J.P.; Keung, E.Z.; Samant, N.S.; Sharma, S.; Natale, C.A.; Terranova, C.J.; Maitituoheti, M.; Amin, S.B.; et al. Systematic Epigenomic Analysis Reveals Chromatin States Associated with Melanoma Progression. Cell Rep. 2017, 19, 875–889.
- Wilmott, J.S.; Colebatch, A.J.; Kakavand, H.; Shang, P.; Carlino, M.S.; Thompson, J.F.; Long, G.V.; Scolyer, R.A.; Hersey, P. Expression of the class 1 histone deacetylases HDAC8 and 3 are associated with improved survival of patients with metastatic melanoma. Mod. Pathol. Off. J. USA Can. Acad. Pathol. Inc. 2015, 28, 884–894.
- Hu, Z.; Rong, Y.; Li, S.; Qu, S.; Huang, S. Upregulated Histone Deacetylase 6 Associates with Malignant Progression of Melanoma and Predicts the Prognosis of Patients. Cancer Manag. Res. 2020, 12, 12993–13001.
- Liu, J.; Gu, J.; Feng, Z.; Yang, Y.; Zhu, N.; Lu, W.; Qi, F. Both HDAC5 and HDAC6 are required for the proliferation and metastasis of melanoma cells. J. Transl. Med. 2016, 14, 7.
- Wilking, M.J.; Singh, C.; Nihal, M.; Zhong, W.; Ahmad, N. SIRT1 deacetylase is overexpressed in human melanoma and its small molecule inhibition imparts anti-proliferative response via p53 activation. Arch. Biochem. Biophys. 2014, 563, 94–100.
- Sun, T.; Jiao, L.; Wang, Y.; Yu, Y.; Ming, L. SIRT1 induces epithelial-mesenchymal transition by promoting autophagic degradation of E-cadherin in melanoma cells article. Cell Death Dis. 2018, 9, 1–10.
- George, J.; Nihal, M.; Singh, C.K.; Zhong, W.; Liu, X.; Ahmad, N. Pro-Proliferative Function of Mitochondrial Sirtuin Deacetylase SIRT3 in Human Melanoma. J. Invest. Dermatol. 2016, 136, 809–818.
- Garcia-Peterson, L.M.; Ndiaye, M.A.; Singh, C.K.; Chhabra, G.; Huang, W.; Ahmad, N. SIRT6 histone deacetylase functions as a potential oncogene in human melanoma. Genes Cancer 2017, 8, 701–712.
- George, J.; Nihal, M.; Singh, C.K.; Ahmad, N. 4′-Bromo-resveratrol, a dual Sirtuin-1 and Sirtuin-3 inhibitor, inhibits melanoma cell growth through mitochondrial metabolic reprogramming. Mol. Carcinog. 2019, 58, 1876–1885.
- Kuźbicki, L.; Lange, D.; Strączyńska-Niemiec, A.; Chwirot, B.W. JARID1B expression in human melanoma and benign melanocytic skin lesions. Melanoma Res. 2013, 23, 8–12.
- Roesch, A.; Becker, B.; Meyer, S.; Wild, P.; Hafner, C.; Landthaler, M.; Vogt, T. Retinoblastoma-binding protein 2-homolog 1: A retinoblastoma-binding protein downregulated in malignant melanomas. Mod. Pathol. Off. J. USA Can. Acad. Pathol. Inc 2005, 18, 1249–1257.
- Chauvistré, H.; Daignault, S.; Shannan, B.; Ju, R.; Picard, D.; Vogel, F.; Egetemaier, S.; Krepler, C.; Rebecca, V.; Sechi, A.; et al. The Janus-faced role of KDM5B heterogeneity in melanoma: Differentiation as a situational driver of both growth arrest and drug-resistance. 2020; preprint.
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594.
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516.
- Long, G.V.; Weber, J.S.; Infante, J.R.; Kim, K.B.; Daud, A.; Gonzalez, R.; Sosman, J.A.; Hamid, O.; Schuchter, L.; Cebon, J.; et al. Overall Survival and Durable Responses in Patients With BRAF V600-Mutant Metastatic Melanoma Receiving Dabrafenib Combined With Trametinib. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 871–878.
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114.
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703.
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93.
- Clark, M.E.; Rizos, H.; Pereira, M.R.; McEvoy, A.C.; Marsavela, G.; Calapre, L.; Meehan, K.; Ruhen, O.; Khattak, M.A.; Meniawy, T.M.; et al. Detection of BRAF splicing variants in plasma-derived cell-free nucleic acids and extracellular vesicles of melanoma patients failing targeted therapy therapies. Oncotarget 2020, 11, 4016–4027.
- Shi, H.; Moriceau, G.; Kong, X.; Lee, M.-K.; Lee, H.; Koya, R.C.; Ng, C.; Chodon, T.; Scolyer, R.A.; Dahlman, K.B.; et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat. Commun. 2012, 3, 724.
- Perna, D.; Karreth, F.A.; Rust, A.G.; Perez-Mancera, P.A.; Rashid, M.; Iorio, F.; Alifrangis, C.; Arends, M.J.; Bosenberg, M.W.; Bollag, G.; et al. BRAF inhibitor resistance mediated by the AKT pathway in an oncogenic BRAF mouse melanoma model. Proc. Natl. Acad. Sci. USA 2015, 112, E536–E545.
- Basile, K.J.; Aplin, A.E. Downregulation of Noxa by RAF/MEK inhibition counteracts cell death response in mutant B-RAF melanoma cells. Am. J. Cancer Res. 2012, 2, 726–735.
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.-K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977.
- Dugo, M.; Nicolini, G.; Tragni, G.; Bersani, I.; Tomassetti, A.; Colonna, V.; Del Vecchio, M.; De Braud, F.; Canevari, S.; Anichini, A.; et al. A melanoma subtype with intrinsic resistance to BRAF inhibition identified by receptor tyrosine kinases gene-driven classification. Oncotarget 2015, 6, 5118–5133.
- Gallagher, S.J.; Tiffen, J.C.; Hersey, P. Histone Modifications, Modifiers and Readers in Melanoma Resistance to Targeted and Immune Therapy. Cancers 2015, 7, 1959–1982.
- Su, Y.; Lu, X.; Li, G.; Liu, C.; Kong, Y.; Lee, J.W.; Ng, R.; Wong, S.; Robert, L.; Warden, C.; et al. Kinetic Inference Resolves Epigenetic Mechanism of Drug Resistance in Melanoma. Cell 2019. preprint.
- Wilting, R.H.; Dannenberg, J.H. Epigenetic mechanisms in tumorigenesis, tumor cell heterogeneity and drug resistance. Drug Resist. Updat. 2012, 15, 21–38.
- Al Emran, A.; Marzese, D.M.; Menon, D.R.; Stark, M.S.; Torrano, J.; Hammerlindl, H.; Zhang, G.; Brafford, P.; Salomon, M.P.; Nelson, N.; et al. Distinct histone modifications denote early stress-induced drug tolerance in cancer. Oncotarget 2018, 9, 8206–8222.
- Hugo, W.; Shi, H.; Sun, L.; Piva, M.; Song, C.; Kong, X.; Moriceau, G.; Hong, A.; Dahlman, K.B.; Johnson, D.B.; et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell 2015, 162, 1271–1285.
- Hammerlindl, H.; Schaider, H. Epigenetics in Melanoma Development and Drug Resistance. In Human Skin Cancers Pathways, Mechanisms, Targets and Treatments; InTech: Tokyo, Japan, 2018.
- Seghers, A.C.; Wilgenhof, S.; Lebbé, C.; Neyns, B. Successful rechallenge in two patients with BRAF-V600-mutant melanoma who experienced previous progression during treatment with a selective BRAF inhibitor. Melanoma Res. 2012, 22, 466–472.
- Atkinson, V.; Batty, K.; Long, G.V.; Carlino, M.S.; Peters, G.D.; Bhave, P.; Moore, M.A.; Xu, W.; Brown, L.J.; Arneil, M.; et al. Activity and safety of third-line BRAF-targeted therapy (TT) following first-line TT and second-line immunotherapy (IT) in advanced melanoma. J. Clin. Oncol. 2020, 38, 10049.
- Cybulska-Stopa, B.; Rogala, P.; Czarnecka, A.M.; Galus, Ł.; Dziura, R.; Rajczykowski, M.; Kubiatowski, T.; Wiśniewska, M.; Gęga-Czarnota, A.; Teterycz, P.; et al. BRAF and MEK inhibitors rechallenge as effective treatment for patients with metastatic melanoma. Melanoma Res. 2020, 30, 465–471.
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80.
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Körbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 2013, 23, 811–825.
- Ravindran Menon, D.; Das, S.; Krepler, C.; Vultur, A.; Rinner, B.; Schauer, S.; Kashofer, K.; Wagner, K.; Zhang, G.; Bonyadi Rad, E.; et al. A stress-induced early innate response causes multidrug tolerance in melanoma. Oncogene 2015, 34, 4448–4459.
- Cartlidge, R.A.; Thomas, G.R.; Cagnol, S.; Jong, K.A.; Molton, S.A.; Finch, A.J.; McMahon, M. Oncogenic BRAF(V600E) inhibits BIM expression to promote melanoma cell survival. Pigment Cell Melanoma Res. 2008, 21, 534–544.
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046.
- Mohana-Kumaran, N.; Hill, D.S.; Allen, J.D.; Haass, N.K. Targeting the intrinsic apoptosis pathway as a strategy for melanoma therapy. Pigment Cell Melanoma Res. 2014, 27, 525–539.
- Tiffen, J.C.; Gunatilake, D.; Gallagher, S.J.; Gowrishankar, K.; Heinemann, A.; Cullinane, C.; Dutton-Regester, K.; Pupo, G.M.; Strbenac, D.; Yang, J.Y.; et al. Targeting activating mutations of EZH2 leads to potent cell growth inhibition in human melanoma by derepression of tumor suppressor genes. Oncotarget 2015, 6, 27023–27036.
- Gallagher, S.J.; Mijatov, B.; Gunatilake, D.; Tiffen, J.C.; Gowrishankar, K.; Jin, L.; Pupo, G.M.; Cullinane, C.; Prinjha, R.K.; Smithers, N.; et al. The epigenetic regulator I-BET151 induces BIM-dependent apoptosis and cell cycle arrest of human melanoma cells. J. Invest. Dermatol. 2014, 134, 2795–2805.
- Heinemann, A.; Cullinane, C.; De Paoli-Iseppi, R.; Wilmott, J.S.; Gunatilake, D.; Madore, J.; Strbenac, D.; Yang, J.Y.; Gowrishankar, K.; Tiffen, J.C.; et al. Combining BET and HDAC inhibitors synergistically induces apoptosis of melanoma and suppresses AKT and YAP signaling. Oncotarget 2015, 6, 21507–21521.
- Neagu, M. Metabolic Traits in Cutaneous Melanoma. Front. Oncol. 2020, 10, 851.
- Witz, I.P. Tumor-microenvironment interactions: Dangerous liaisons. Adv. Cancer Res. 2008, 100, 203–229.
- Falcone, I.; Conciatori, F.; Bazzichetto, C.; Ferretti, G.; Cognetti, F.; Ciuffreda, L.; Milella, M. Tumor Microenvironment: Implications in Melanoma Resistance to Targeted Therapy and Immunotherapy. Cancers 2020, 12, 2870.
- Mazurkiewicz, J.; Simiczyjew, A.; Dratkiewicz, E.; Ziętek, M.; Matkowski, R.; Nowak, D. Stromal Cells Present in the Melanoma Niche Affect Tumor Invasiveness and Its Resistance to Therapy. Int. J. Mol. Sci. 2021, 22, 529.
- Georgescu, S.R.; Tampa, M.; Mitran, C.I.; Mitran, M.I.; Caruntu, C.; Caruntu, A.; Lupu, M.; Matei, C.; Constantin, C.; Neagu, M. Tumour Microenvironment in Skin Carcinogenesis. Adv. Exp. Med. Biol. 2020, 1226, 123–142.
- Passarelli, A.; Mannavola, F.; Stucci, L.S.; Tucci, M.; Silvestris, F. Immune system and melanoma biology: A balance between immunosurveillance and immune escape. Oncotarget 2017, 8, 106132–106142.
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019, 234, 8509–8521.
- Liu, Y.; Guo, J.; Huang, L. Modulation of tumor microenvironment for immunotherapy: Focus on nanomaterial-based strategies. Theranostics 2020, 10, 3099–3117.
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155.
- Turnis, M.E.; Andrews, L.P.; Vignali, D.A.A. Inhibitory receptors as targets for cancer immunotherapy. Eur. J. Immunol. 2015, 45, 1892–1905.
- Yervoy (ipilimumab) FDA Approval History Drugs.com. Available online: https://www.drugs.com/history/yervoy.html (accessed on 17 February 2021).
- Opdivo (nivolumab) FDA Approval History Drugs.com. Available online: https://www.drugs.com/history/opdivo.html (accessed on 17 February 2021).
- Asher, N.; Ben-Betzalel, G.; Lev-Ari, S.; Shapira-Frommer, R.; Steinberg-Silman, Y.; Gochman, N.; Schachter, J.; Meirson, T.; Markel, G. Real World Outcomes of Ipilimumab and Nivolumab in Patients with Metastatic Melanoma. Cancers 2020, 12, 2329.
- Nobel Prize Awarded to Cancer Immunotherapy Researchers. Available online: https://www.cancer.org/latest-news/nobel-prize-awarded-to-cancer-immunotherapy-researchers.html (accessed on 17 February 2021).
- Emran, A.A.; Chatterjee, A.; Rodger, E.J.; Tiffen, J.C.; Gallagher, S.J.; Eccles, M.R.; Hersey, P. Targeting DNA Methylation and EZH2 Activity to Overcome Melanoma Resistance to Immunotherapy. Trends Immunol. 2019, 40, 328–344.
- Lee, P.P.; Fitzpatrick, D.R.; Beard, C.; Jessup, H.K.; Lehar, S.; Makar, K.W.; Pérez-Melgosa, M.; Sweetser, M.T.; Schlissel, M.S.; Nguyen, S.; et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 2001, 15, 763–774.
- Scanlon, S. DNA methylation makes for tired T cells. Science. 2017, 357, 367–368.
- Xiao, Q.; Nobre, A.; Piñeiro, P.; Berciano-Guerrero, M.-Á.; Alba, E.; Cobo, M.; Lauschke, V.M.; Barragán, I. Genetic and Epigenetic Biomarkers of Immune Checkpoint Blockade Response. J. Clin. Med. 2020, 9, 286.
- Fietz, S.; Zarbl, R.; Niebel, D.; Posch, C.; Brossart, P.; Gielen, G.H.; Strieth, S.; Pietsch, T.; Kristiansen, G.; Bootz, F.; et al. CTLA4 promoter methylation predicts response and progression-free survival in stage IV melanoma treated with anti-CTLA-4 immunotherapy (ipilimumab). Cancer Immunol. Immunother. 2020.
- Madore, J.; Vilain, R.E.; Menzies, A.M.; Kakavand, H.; Wilmott, J.S.; Hyman, J.; Yearley, J.H.; Kefford, R.F.; Thompson, J.F.; Long, G.V.; et al. PD-L1 expression in melanoma shows marked heterogeneity within and between patients: Implications for anti-PD-1/PD-L1 clinical trials. Pigment Cell Melanoma Res. 2015, 28, 245–253.
- Sznol, M.; Chen, L. Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer. Clin. cancer Res. an Off. J. Am. Assoc. Cancer Res. 2013, 19, 1021–1034.
- Eddy, K.; Chen, S. Overcoming Immune Evasion in Melanoma. Int. J. Mol. Sci. 2020, 21, 8984.
- Madore, J.; Strbenac, D.; Vilain, R.; Menzies, A.M.; Yang, J.Y.H.; Thompson, J.F.; Long, G.V.; Mann, G.J.; Scolyer, R.A.; Wilmott, J.S. PD-L1 Negative Status is Associated with Lower Mutation Burden, Differential Expression of Immune-Related Genes, and Worse Survival in Stage III Melanoma. Clin. cancer Res. an Off. J. Am. Assoc. Cancer Res. 2016, 22, 3915–3923.
- Micevic, G.; Thakral, D.; McGeary, M.; Bosenberg, M.W. PD-L1 methylation regulates PD-L1 expression and is associated with melanoma survival. Pigment Cell Melanoma Res. 2019, 32, 435–440.
- Fröhlich, A.; Sirokay, J.; Fietz, S.; Vogt, T.J.; Dietrich, J.; Zarbl, R.; Florin, M.; Kuster, P.; Saavedra, G.; Valladolid, S.R.; et al. Molecular, clinicopathological, and immune correlates of LAG3 promoter DNA methylation in melanoma. EBioMedicine 2020, 59, 102962.
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986.
- Zingg, D.; Arenas-Ramirez, N.; Sahin, D.; Rosalia, R.A.; Antunes, A.T.; Haeusel, J.; Sommer, L.; Boyman, O. The Histone Methyltransferase Ezh2 Controls Mechanisms of Adaptive Resistance to Tumor Immunotherapy. Cell Rep. 2017, 20, 854–867.
- Tiffen, J.; Wilson, S.; Gallagher, S.J.; Hersey, P.; Filipp, F. V Somatic Copy Number Amplification and Hyperactivating Somatic Mutations of EZH2 Correlate With DNA Methylation and Drive Epigenetic Silencing of Genes Involved in Tumor Suppression and Immune Responses in Melanoma. Neoplasia 2016, 18, 121–132.
- Chen, B.-F.; Chan, W.-Y. The de novo DNA methyltransferase DNMT3A in development and cancer. Epigenetics 2014, 9, 669–677.
- Badeaux, A.I.; Shi, Y. Emerging roles for chromatin as a signal integration and storage platform. Nat. Rev. Mol. Cell Biol. 2013, 14, 211–224.
- Schlesinger, Y.; Straussman, R.; Keshet, I.; Farkash, S.; Hecht, M.; Zimmerman, J.; Eden, E.; Yakhini, Z.; Ben-Shushan, E.; Reubinoff, B.E.; et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 2007, 39, 232–236.

