 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jan O. Aaseth | + 1612 word(s) | 1612 | 2021-09-09 04:42:04 | | | |
| 2 | Dean Liu | Meta information modification | 1612 | 2021-09-13 03:29:59 | | |
Video Upload Options
Glycation has been shown to induce aggregation of a wide spectrum of proteins including those implicated in the pathogenesis of AD. A recent in vitro fluorescence study revealed a significant impact of glucose levels on Aβ1-40 aggregation, resulting in additional types of formed oligomers. Correspondingly, Aβ glycation was associated with aggravation of amyloid neurotoxicity through up-regulation of RAGE and subsequent GSK-3 activation.
1. Introduction
Alzheimer’s disease (AD) is a neurodegenerative condition of high prevalence in older age. In developed countries almost 10% of people over the age of 65 suffer from AD [1] and AD prevalence is increasing globally. During the upcoming decades AD will contribute to an increased socio-economic burden and severe suffering for patients and caregivers. Against this background, measures to modify the serious course of AD are urgently needed.
Most AD patients suffer from the sporadic form, the genetic AD accounting for less than 2% of diagnosed cases [2]. Many sporadic AD patients (almost 25%) are carriers of the ApoE4 allele of apolipoprotein E (chromosome 19) [3]. The mechanisms by which the ApoE4 allele contributes to AD and to increased levels of amyloid beta (Aβ) are unknown, yet the same allele appears to be related to telomere shortening and accelerated aging [4]. It is known that the import of cholesterol into the neurons is achieved by apolipoprotein E (ApoE) via receptors low-density lipoprotein receptor-related protein1 and 8 (LRP1 and LRP8) on the cell surface. It has been suggested that a co-causal factor for AD is changed receptor function of LRP1 or LRP8, and that an underlying cause is inappropriate neuronal supply of cholesterol combined with dysmetabolism of the copper chaperone APP (amyloid precursor protein) [5]. In particular, the essential trace element selenium (Se) is also transported as selenoprotein P (SELENOP) to the brain via a low-density lipoprotein receptor, viz. the LRP8 receptor [6]. In addition, another lipoprotein receptor (LRP1) is implicated in the clearance of Aβ from the brain to the peripheral circulation [7]. Interestingly, this clearance appears to be dependent on the ApoE isoform, as carriers of the apoE4 allele exert reduced efflux of Aβ from the brain, as assessed in in vitro models [8]. Combined with aging hypertension, dyslipidemia and metabolic syndrome (MetS) are risk factors for developing sporadic AD [9].
Language problems and declining recall of recent events are early AD characteristics [10]. These symptoms are attributed to dysfunctions in the hippocampal area of the temporal lobe [11]. Degenerations develop gradually in both the temporal and parietal regions of the brain. These changes are accompanied by a deterioration of hippocampal cholinergic signaling [12]. In clinical medicine, acetylcholinesterase inhibitors have been adopted to alleviate synaptic dysfunction in AD, acting by increasing acetylcholine levels at synaptic loci, however without arresting the disease progression [13]. Symptomatic relief is also achieved by memantine, a compound that acts by blocking the functional dominance of the transmitter N-methyl-aspartate [14]. However, transmitter dysfunctions observed in AD patients are not considered the initiating events in the pathogenesis. Biochemical events initiating AD development most probably reside in some basic impairments of vital intracellular proteins, e.g., the microtubule-associated proteins responsible for the neuronal integrity.
2. On Traditional Hypotheses for AD Etiology
However, in AD, a less soluble variant, the Aβ-peptide with 42 amino acids, usually referred to as Aβ, is formed in excess and makes up the amyloid core in the characteristic precipitated plaques [15][16].
Pyroglutamate-modified Aβ peptides that are formed upon glutaminyl cyclase catalysis are strongly associated with AD [17]. Therefore, antibodies to pyroglutamate-modified Aβ peptides as well as glutaminyl cyclase inhibitors may be considered as an additional immunotherapeutic approach to AD [17].
The ubiquitin-proteasome system in neurons appears to be overloaded and deranged in AD, ultimately leading to cell death. Oxidized proteins contribute to a pathologic overloading in aging. Cell deterioration and death lead to extracellular escape of Aβ deposits. Age-related mitochondrial dysfunctions may accelerate the formation of protein oxidation products [18].
Accumulation of damaged mitochondria is common in brain tissues from AD patients and in AD animal models, in addition to autophagosomes, which seem to be formed at mitochondrial endoplasmic reticulum contact sites (MERCS) [19] together with Aβ plaques [20]. It has been hypothesized that formation of abnormal MERCS in AD might lead to mitochondrial dysfunction due to an influx of calcium into mitochondria from the endoplasmic reticulum and Aβ aggregates blocking mitochondrial export of Ca ++ , in addition to dysfunctional autophagosome synthesis [21]. Physiologically, the major pathway for removal of damaged mitochondria is the ATP-dependent ubiquitin proteasome pathway in addition to mitophagy. A tempting hypothesis is that, together with the overloaded proteasome pathway, a defective mitophagy may play a role in the AD etiopathogenesis [22].
3. Hypothesis Involving Metabolic Syndrome and Glycation
High levels of glucose in diabetes mellitus, as well as monosaccharides such as fructose and glyceraldehyde, react non-enzymatically with sulfhydryl groups of proteins, lipids and nucleic acids, leading to the formation of advanced glycation end-products (AGEs). Briefly, protein glycation is a series of non-enzymatic reactions collectively termed the Maillard reaction [23] ( Figure 1 ). At the first step, reaction between the protein free amino group and carbonyl group of a reducing carbohydrate (glucose) results in formation of a Schiff base that is subsequently rearranged to a more stable Amadori product. The latter undergoes a series of rearrangement, oxidation and dehydration reactions in the formation of AGEs. Another mechanism for AGE formation involves generation of reactive carbonyl species (glyoxal, methylglyoxal, 3-desoxyglucosone) that interact with amino acid residues of proteins resulting in AGE formation [24].
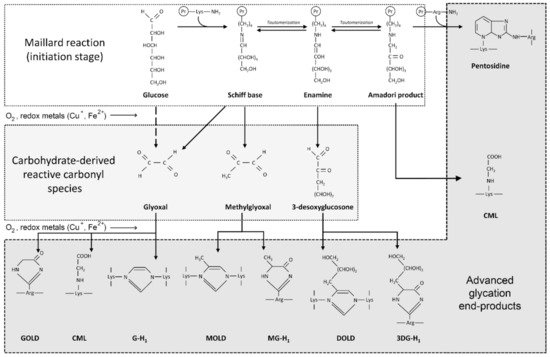
Irreversible modification of biological macromolecules during AGE formation results in alteration of its structure and function, and AGEs also possess toxic properties [25]. Glycation has been shown to induce aggregation of a wide spectrum of proteins including those implicated in the pathogenesis of AD [23]. A recent in vitro fluorescence study revealed a significant impact of glucose levels on Aβ1-40 aggregation, resulting in additional types of formed oligomers [26]. Correspondingly, Aβ glycation was associated with aggravation of amyloid neurotoxicity through up-regulation of RAGE and subsequent GSK-3 activation [27].
The role of AGEs in AD may be mediated by up-regulation of the receptor for advanced glycation end-products (RAGE). RAGE is a transmembrane protein of the immunoglobulin superfamily that in parallel with AGE interacts with multiple ligands including high-mobility group protein (B)1, S100 protein and Aβ, to name a few. Activated RAGE is involved in a variety of processes including inflammation, oxidative stress, apoptosis, autophagy, proliferation and migration [28]. Correspondingly, RAGE is considered to play a significant role in the pathogenesis of AD [29].
As for the mechanisms of the role of RAGE in neuroinflammation, recent studies have demonstrated that RAGE activation may activate ERK1/2, JNK and p38 MAPK signaling, as well as ROS overproduction through NAPDH oxidase, with subsequent induction of the NF-kB pathway [30]. In turn, NF-kB activation was also shown to up-regulate RAGE expression, thus maintaining the vicious circle of neuroinflammation [31].
4. The Role of Copper (Cu) and Iron (Fe) in Alzheimer’s Disease
The association of Fe with AD pathology is supported by the finding that high ferritin levels in cerebrospinal fluid (CSF) correlated with the transition of mild cognitive impairment (MCI) into AD, as observed in a cohort study [32]. Furthermore, CSF ferritin levels were strongly associated with cognitive decline in carriers of the ApoE4 allele [33]. Of interest is also the observation that the binding of Fe and Cu ions to phosphorylated tau protein precedes the formation of intracellular tangles [34]. It is also known that cations of these same transition elements accumulate in AD plaques enhancing the progression of the Aβ cascade [35].
Masaldan and colleagues have suggested that the complex pathological process of AD can be described as a ferroptosis [36], underscoring a role of iron interactions in addition to lipid dysmetabolism in the generation of the characteristic AD pathology.
Taken together, these data demonstrate that the redox metals Cu and Fe are involved in the complex AD pathogenesis. Given their role in oxidative stress, as well as the earlier discussed involvement in glycation processes and AGE/RAGE signaling in molecular mechanisms of AD, we propose that metals may play a significant role in AD etiology secondary to their impact on AGE generation and subsequent toxicity.
Several studies have addressed the role of iron in AGE formation, which may also underlie the potential involvement of Fe 2+ in neurodegeneration. Specifically, the potential catalytic role of Fe 2+ in the formation of AGEs in type 1 collagen has been clearly demonstrated [37]. In patients with β-thalassemia major, serum iron and especially non-transferrin-bound iron are typically elevated, and a positive correlation has been reported between this elevation and AGE (carboxymethyl-lysine and pentosidine) concentrations [38]. Correspondingly, in our recent experimental study Fe supplementation in obese rats induced a significant accumulation of AGEs and especially of CML in the liver [39]. Iron (Fe 3+ ) is capable of inducing formation of DNA-AGE adducts, especially when combined with glyoxal and arginine [40]. However, direct evidence demonstrating the trilateral relationship between iron, AGE formation and Aβ accumulation and/or toxicity are lacking.
References
- Alzheimer’s Association. 2021 Alzheimers disease facts and figures. Alzheimers Dement. 2021, 17, 327–406.
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339.
- Sadigh-Eteghad, S.; Talebi, M.; Farhoudi, M. Association of apolipoprotein E epsilon 4 allele with sporadic late onset Alzheimer’s disease. A meta-analysis. Neurosciences 2012, 17, 321–326.
- Jacobs, E.G.; Kroenke, C.; Lin, J.; Epel, E.S.; Kenna, H.A.; Blackburn, E.H.; Rasgon, N.L. Accelerated cell aging in female APOE-epsilon4 carriers: Implications for hormone therapy use. PLoS ONE 2013, 8, e54713.
- Liu, Q.; Zerbinatti, C.V.; Zhang, J.; Hoe, H.S.; Wang, B.; Cole, S.L.; Herz, J.; Muglia, L.; Bu, G. Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron 2007, 56, 66–78.
- Schweizer, U.; Bohleber, S.; Zhao, W.; Fradejas-Villar, N. The Neurobiology of Selenium: Looking Back and to the Future. Front. Neurosci 2021, 15, 652099.
- Deane, R.; Bell, R.D.; Sagare, A.; Zlokovic, B.V. Clearance of amyloid-beta peptide across the blood-brain barrier: Implication for therapies in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2009, 8, 16–30.
- Bachmeier, C.; Paris, D.; Beaulieu-Abdelahad, D.; Mouzon, B.; Mullan, M.; Crawford, F. A multifaceted role for apoE in the clearance of beta-amyloid across the blood-brain barrier. Neurodegener. Dis. 2013, 11, 13–21.
- Drachman, D.A. The amyloid hypothesis, time to move on: Amyloid is the downstream result, not cause, of Alzheimer’s disease. Alzheimers Dement. 2014, 10, 372–380.
- Jahn, H. Memory loss in Alzheimer’s disease. Dialogues Clin. Neurosci. 2013, 15, 445–454.
- Lim, H.K.; Jung, W.S.; Ahn, K.J.; Won, W.Y.; Hahn, C.; Lee, S.Y.; Kim, I.; Lee, C.U. Relationships between hippocampal shape and cognitive performances in drug-naive patients with Alzheimer’s disease. Neurosci. Lett. 2012, 516, 124–129.
- Konishi, K.; Hori, K.; Tani, M.; Tomioka, H.; Kitajima, Y.; Akashi, N.; Inamoto, A.; Kurosawa, K.; Yuda, H.; Hanashi, T.; et al. Hypothesis of Endogenous Anticholinergic Activity in Alzheimer’s Disease. Neurodegener. Dis. 2015, 15, 149–156.
- Wallace, T.L.; Bertrand, D. Importance of the nicotinic acetylcholine receptor system in the prefrontal cortex. Biochem. Pharm. 2013, 85, 1713–1720.
- Parsons, C.G.; Stoffler, A.; Danysz, W. Memantine: A NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system—Too little activation is bad, too much is even worse. Neuropharmacology 2007, 53, 699–723.
- Williams, T.L.; Serpell, L.C. Membrane and surface interactions of Alzheimer’s Abeta peptide—Insights into the mechanism of cytotoxicity. FEBS J. 2011, 278, 3905–3917.
- Warmlander, S.; Tiiman, A.; Abelein, A.; Luo, J.; Jarvet, J.; Soderberg, K.L.; Danielsson, J.; Graslund, A. Biophysical studies of the amyloid beta-peptide: Interactions with metal ions and small molecules. ChemBioChem 2013, 14, 1692–1704.
- Perez-Garmendia, R.; Gevorkian, G. Pyroglutamate-Modified Amyloid Beta Peptides: Emerging Targets for Alzheimer’s Disease Immunotherapy. Curr. Neuropharmacol. 2013, 11, 491–498.
- Andreyev, A.Y.; Kushnareva, Y.E.; Murphy, A.N.; Starkov, A.A. Mitochondrial ROS Metabolism: 10 Years Later. Biochemistry 2015, 80, 517–531.
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122.
- Nilsson, P.; Loganathan, K.; Sekiguchi, M.; Matsuba, Y.; Hui, K.; Tsubuki, S.; Tanaka, M.; Iwata, N.; Saito, T.; Saido, T.C. Abeta secretion and plaque formation depend on autophagy. Cell Rep. 2013, 5, 61–69.
- Leal, N.S.; Martins, L.M. Mind the Gap: Mitochondria and the Endoplasmic Reticulum in Neurodegenerative Diseases. Biomedicines 2021, 9, 227.
- Fang, E.F. Mitophagy and NAD+ inhibit Alzheimer disease. Autophagy 2019, 15, 1112–1114.
- Chaudhuri, J.; Bains, Y.; Guha, S.; Kahn, A.; Hall, D.; Bose, N.; Gugliucci, A.; Kapahi, P. The Role of Advanced Glycation End Products in Aging and Metabolic Diseases: Bridging Association and Causality. Cell Metab. 2018, 28, 337–352.
- Brings, S.; Fleming, T.; Freichel, M.; Muckenthaler, M.U.; Herzig, S.; Nawroth, P.P. Dicarbonyls and Advanced Glycation End-Products in the Development of Diabetic Complications and Targets for Intervention. Int. J. Mol. Sci. 2017, 18, 984.
- Rungratanawanich, W.; Qu, Y.; Wang, X.; Essa, M.M.; Song, B.J. Advanced glycation end products (AGEs) and other adducts in aging-related diseases and alcohol-mediated tissue injury. Exp. Mol. Med. 2021, 53, 168–188.
- Alghamdi, A.; Forbes, S.; Birch, D.J.S.; Vyshemirsky, V.; Rolinski, O.J. Detecting beta-amyloid glycation by intrinsic fluorescence—Understanding the link between diabetes and Alzheimer’s disease. Arch. Biochem. Biophys. 2021, 704, 108886.
- Li, X.H.; Du, L.L.; Cheng, X.S.; Jiang, X.; Zhang, Y.; Lv, B.L.; Liu, R.; Wang, J.Z.; Zhou, X.W. Glycation exacerbates the neuronal toxicity of beta-amyloid. Cell Death Dis. 2013, 4, e673.
- Lee, E.J.; Park, J.H. Receptor for Advanced Glycation Endproducts (RAGE), Its Ligands, and Soluble RAGE: Potential Biomarkers for Diagnosis and Therapeutic Targets for Human Renal Diseases. Genom. Inf. 2013, 11, 224–229.
- Cai, Z.; Liu, N.; Wang, C.; Qin, B.; Zhou, Y.; Xiao, M.; Chang, L.; Yan, L.J.; Zhao, B. Role of RAGE in Alzheimer’s Disease. Cell Mol. Neurobiol. 2016, 36, 483–495.
- Ray, R.; Juranek, J.K.; Rai, V. RAGE axis in neuroinflammation, neurodegeneration and its emerging role in the pathogenesis of amyotrophic lateral sclerosis. Neurosci. Biobehav. Rev. 2016, 62, 48–55.
- Tobon-Velasco, J.C.; Cuevas, E.; Torres-Ramos, M.A. Receptor for AGEs (RAGE) as mediator of NF-kB pathway activation in neuroinflammation and oxidative stress. CNS Neurol. Disord. Drug Targets 2014, 13, 1615–1626.
- Ayton, S.; Faux, N.G.; Bush, A.I.; Alzheimer’s Disease Neuroimaging Initiative. Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE. Nat. Commun. 2015, 6, 6760.
- Ayton, S.; Faux, N.G.; Bush, A.I. Association of Cerebrospinal Fluid Ferritin Level with Preclinical Cognitive Decline in APOE-epsilon4 Carriers. JAMA Neurol. 2017, 74, 122–125.
- Ahmadi, S.; Zhu, S.; Sharma, R.; Wilson, D.J.; Kraatz, H.B. Interaction of metal ions with tau protein. The case for a metal-mediated tau aggregation. J. Inorg. Biochem. 2019, 194, 44–51.
- Bourassa, M.W.; Leskovjan, A.C.; Tappero, R.V.; Farquhar, E.R.; Colton, C.A.; Van Nostrand, W.E.; Miller, L.M. Elevated copper in the amyloid plaques and iron in the cortex are observed in mouse models of Alzheimer’s disease that exhibit neurodegeneration. Biomed. Spectrosc. Imaging 2013, 2, 129–139.
- Masaldan, S.; Bush, A.I.; Devos, D.; Rolland, A.S.; Moreau, C. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radic. Biol. Med. 2019, 133, 221–233.
- Xiao, H.; Cai, G.; Liu, M. Fe2+-catalyzed non-enzymatic glycosylation alters collagen conformation during AGE-collagen formation in vitro. Arch. Biochem. Biophys. 2007, 468, 183–192.
- Mirlohi, M.S.; Yaghooti, H.; Shirali, S.; Aminasnafi, A.; Olapour, S. Increased levels of advanced glycation end products positively correlate with iron overload and oxidative stress markers in patients with beta-thalassemia major. Ann. Hematol. 2018, 97, 679–684.
- Chen, S.H.; Yuan, K.C.; Lee, Y.C.; Shih, C.K.; Tseng, S.H.; Tinkov, A.A.; Skalny, A.V.; Chang, J.S. Iron and Advanced Glycation End Products: Emerging Role of Iron in Androgen Deficiency in Obesity. Antioxidants 2020, 9, 261.
- Shahab, U.; Tabrez, S.; Khan, M.S.; Akhter, F.; Khan, M.S.; Saeed, M.; Ahmad, K.; Srivastava, A.K.; Ahmad, S. Immunogenicity of DNA-advanced glycation end product fashioned through glyoxal and arginine in the presence of Fe3+: Its potential role in prompt recognition of diabetes mellitus auto-antibodies. Chem. Biol. Interact. 2014, 219, 229–240.

