Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kerstin Borgmann | + 3051 word(s) | 3051 | 2021-08-26 09:42:45 | | | |
| 2 | Vivi Li | Meta information modification | 3051 | 2021-09-08 11:42:09 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Borgmann, K. Cancer Stem Cells and Radioresistance. Encyclopedia. Available online: https://encyclopedia.pub/entry/14000 (accessed on 26 June 2026).
Borgmann K. Cancer Stem Cells and Radioresistance. Encyclopedia. Available at: https://encyclopedia.pub/entry/14000. Accessed June 26, 2026.
Borgmann, Kerstin. "Cancer Stem Cells and Radioresistance" Encyclopedia, https://encyclopedia.pub/entry/14000 (accessed June 26, 2026).
Borgmann, K. (2021, September 08). Cancer Stem Cells and Radioresistance. In Encyclopedia. https://encyclopedia.pub/entry/14000
Borgmann, Kerstin. "Cancer Stem Cells and Radioresistance." Encyclopedia. Web. 08 September, 2021.
Copy Citation
The current preclinical and clinical findings demonstrate that, in addition to the conventional clinical and pathological indicators that have a prognostic value in radiation oncology, the number of cancer stem cells (CSCs) and their inherent radioresistance are important parameters for local control after radiotherapy.
cancer stem cells
DNA repair
radioresistance
5Rs of radiation biology
1. Introduction
According to the global cancer statistics, cancer incidence and mortality are increasing worldwide with an estimated 29 million new cancer cases by 2030 [1][2][3][4], attributed to constant growth and aging of the human population [5]. This rapid growth of cancer rates suggests that improving diagnosis of cancer and integration of individualized treatment into the mainstream clinical practice are of utmost importance. As of now, about half of all cancer patients receive radiation therapy as a treatment modality during the history of disease [6]. The recent improvements in radiation technologies and delivery substantially increased efficiency and quality of treatment [6]. Nevertheless, one of the fundamental problems of radiation oncology is tumor resistance to radiation doses which cause an acceptable degree of normal tissue toxicity. Tumor radioresistance leads to loco-regional control failure and disease progression [7][8]. The curative potential of radiotherapy depends on its ability to cause a reproductive death of tumor cells via accumulation of non-repairable DNA lesions, thereby removing cancer cells from the clonogenic pool [9][10][11][12].
In the last few decades, studies in different fields of the biological sciences demonstrated that cancer cells are heterogeneous in their tumor-initiating properties which contribute to tumor growth and metastasis development [10][13][14][15]. Tumor cell transplantation experiments, first in isogenic murine models by Hewitt and Wilson and then in human xenograft tumor models, demonstrated that the therapeutic potential of radiotherapy is defined by the number of tumorigenic cells in these experimental tumors [9][12][16][17][18]. These findings are in agreement with clinical data for patients treated with radiotherapy; smaller tumor size was associated with longer patient survival because larger tumors might contain higher absolute number of tumorigenic cells. Together with other clinical and pathological parameters, tumor size is used for radiation therapy patient stratification [19][20].
Identification of markers for the detection of tumor-initiating cells, first in leukemia by the group of John Dick (CD (cluster of differentiation) 34+/CD38−) and then in solid tumors including glioma (CD133+, aldehyde dehydrogenase, ALDH1+), breast (epithelial cell adhesion molecule, EpCAM+/CD44high/CD24low, ALDH1+), colorectal (CD133+, EpCAMhigh/CD44+, ALDH1+), head and neck squamous carcinoma (CD44+), and other cancers as discussed elsewhere [21], paved the way for isolating, enriching, and analyzing these tumorigenic cells in different tumor entities [15][21][22]. These findings indicated that tumor-initiating cells, also called cancer stem cells (CSCs) possess two fundamental properties that make them different from other tumor cells; they have unlimited capacity to self-renew (e.g., divide asymmetrically to produce an identical copy of itself and more differentiated progeny cells) and differentiate to all cell populations present in the original tumors. These properties make these cells a root of tumor growth and recurrence and, thus, an important marker for tumor diagnosis, prognosis, and treatment, as well as critical targets for cancer therapy. Strong evidence is emerging to support the dynamic nature of tumor stemness which can be influenced by genetic alterations, epigenetic reprogramming and the tumor microenvironment [13][23]. Although tumor cell heterogeneity displays a much higher level of complexity than can be explained by the hierarchical model, CSC populations remain critical targets and biomarkers for cancer treatments.
2. Cancer Stem Cells and 5Rs of Radiation Biology
Radiation therapy is one of the key anti-cancer treatment options along with surgery and chemotherapy [6]. However, a high inter- and intratumoral variability in the phenotypical and functional properties of cancer cells remains the major obstacle to improving cancer survival rates, which is especially true for patients with locally advanced tumors [15][24], and reliable biomarkers for patient stratification are of high demand [25]. As of today, only few biological parameters are suggested as potential biomarkers for radiation oncology including tumor size, hypoxia, or positivity for human papilloma virus (HPV) for head and neck squamous cell carcinoma (HNSCC) [11]. The biology-based patient stratification aims to select potential responders and non-responders to increase the probability of cancer cure by radiation therapy. It is based on the radiobiological concept of the “5Rs” which are repair, redistribution, repopulation, reoxygenation, and intrinsic radioresistance [26][27].
The curative potential of radiotherapy depends on the scale and quality of DNA damage in the exposed tumor tissues. Ionizing radiation induces different types of DNA damage with DNA double-strand breaks (DSBs) as major lethal DNA lesions. If the levels of radiation-induced DSBs exceed the DNA repair capacity of tumor cells, this might result in cell cycle arrest, tumor cell senescence, and death. The cell fate decision following radiation exposure depends on the amount of critical DNA damage. A low level of DNA lesions triggers DNA repair mechanisms and DNA damage checkpoints, which arrest cell cycle progression in the presence of DNA damage and allow cells to repair DNA before returning to the proliferative pool [28]. However, if the amount of DNA damage is unrepairable, the cells activate death programs [28][29]. The first “R” refers to DNA repair as one of the key determinants of tumor cell survival after radiation therapy. The cellular response to DNA damage is a complex process including activation of the DNA damage response pathways and DNA repair mechanisms as reviewed elsewhere [28][30]. In addition to direct DNA ionization, radiation-induced DNA damage is caused by highly chemically reactive species produced by radiolysis of water and by disruption of normal mitochondrial functions [30]. These chemically active molecules, called reactive oxygen species (ROS) and reactive nitrogen species (RNS), induce damage of DNA, protein oxidation, and lipid peroxidation, and might cause cell senescence and death [31]. These chemical species are regular by-products of cell metabolism and important second messengers [31][32]. Under physiological conditions, the intracellular concentration of ROS is maintained by the scavenging system which includes, e.g., glutathione, thioredoxin, and enzymatic proteins thioredoxin reductase, dismutase, peroxidase, and catalase, all of which might also shield the cells against radiation-induced oxidative stress [33].
Accumulating preclinical evidence indicates that many of these protective mechanisms are activated in CSC populations possibly resulting in treatment resistance (Figure 1). These findings also suggest that signaling pathways that control DNA integrity and repair in CSCs may serve as a promising target in cancer therapy, as discussed in the next sections of this review.
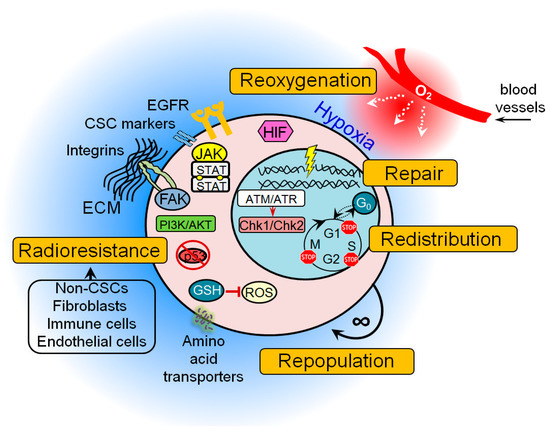
Figure 1. The general mechanisms of cancer stem cell (CSC) radioresistance. Many protective mechanisms are activated in CSCs that result in treatment resistance. These include intrinsic determinants (activation of the pro-survival pathways, enhanced DNA repair capability, protection against oxidative stress, unlimited self-renewal potential, impaired cell cycle arrest, and apoptosis) and extrinsic determinants such as hypoxic microenvironment and a protective CSC niche consisting of the cellular components (e.g., non-CSCs, fibroblasts, immune cells, endothelial cells), soluble factors (e.g., growth factors, hormones and cytokines), and extracellular matrix. ATM—ataxia–telangiectasia mutated; ATR—ATM- and Rad3-Related; Chk1—checkpoint kinase 1; Chk2—checkpoint kinase 2; ECM—extracellular matrix; FAK—focal adhesion kinase; GSH—glutathione; JAK—Janus kinase; EGFR—epidermal growth factor receptor; HIF—hypoxia-inducible factor; PI3K—phosphatidylinositol 3-kinases; ROS—reactive oxygen species.
To repair DSBs, cells employ two major mechanisms: the more error-prone non-homologous end joining (NHEJ) and the more accurate homologous recombination (HR). In addition, tumor cells also have two extremely error-prone DSB back-up repair mechanisms for both NHEJ and HR, the alt-EJ and single-strand annealing (SSA) [34]. In mammalian cells, HR occurs only in the late synthesis (S) phase and less in the gap 2 (G2) phase of the cell cycle when the DNA template on the sister chromatid is available for recombination, whereas NHEJ is active throughout the entire cell cycle with highest efficiency during the G2/mitosis (M) stage and is predominant in G0, G1, and early S phases [35][36][37][38][39].
An activation of these different DNA repair mechanisms at specific phases of the cell cycle results in differences in radiosensitivity throughout the cell cycle, with increased radioresistance in the late S phase and increased radioresponsiveness in G2 and M phases [40]. Increased cell radioresistance in the S phase was attributed to an increased level of DNA replication enabling the HR process [40]. Resistance caused by HR-mediated repair is further enhanced by the presence of all available DNA repair pathways, including those that go beyond the repair of DSBs [39][41]. Therefore, the second “R” refers to redistribution of tumor cells in the cell cycle during radiotherapy to the more radiosensitive cell states. Similar to normal stem cells, some CSC populations are slow-proliferating or quiescent cells [42]. A comparative analysis of DNA repair in the proliferating and quiescent tissues showed that quiescent cells lack DNA repair efficiency and display sustained accumulation of DSBs that could cause an activation of p53 signaling and apoptosis [43][44]. In contrast to the normal tissues and tumor bulk, CSCs have attenuated activation of p53 after radiation-induced DNA damage and, as result, impaired cell cycle arrest and apoptosis that, in the long run, might lead to accumulation of DNA mutations [15][44]. This accumulation of mutation burden over time increases intratumoral heterogeneity and leads to tumor evolution and disease progression [15]. The CSC evolution during tumor development and treatment is associated with activation of different pro-survival pathways which cause intrinsic radioresistance of tumor cells such as epidermal growth factor receptor (EGFR), phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR), wingless-type MMTV integration site family (WNT), Notch, and Hedgehog signaling [11][14][45], and different agents targeting these molecular pathways are currently in preclinical and clinical development.
In addition to the intrinsic mechanisms of radioresistance, the fate of tumor cells after radiotherapy depends on the plethora of cues coming from the tumor microenvironment. Tumors show growth comparable to that of rapidly proliferating tissues with a comparably high demand for oxygen and nutrients from the blood vessels. A number of recent studies demonstrated that tumors can make their own blood vessels and some CSC populations, e.g., in glioblastoma, are capable of differentiating into endothelial cells [46][47]. However, sometimes tumors outgrow their vasculature and develop regions with an inadequate nourishment and supply of oxygen. The hypoxic microenvironment might promote radiotherapy resistance by decreasing DSB burden mediated by oxygen-dependent free-radical production as discussed elsewhere [48][49]. Hypoxia induces significant changes in tumor metabolism due to the deficiency of nutrients, low concentration of oxygen, and deregulation of transporter proteins and metabolic enzymes [49]. An impaired mitochondrial respiration in tumor cells under hypoxia increases glucose uptake to cover high energetic demands of growing tumors. This biological feature is used in clinical practice for tumor imaging by positron emission tomography (PET) using radiolabeled glucose analog fluorine-18-2-fluoro-2-deoxy-d-glucose (18F-FDG) [50][51]. Sustained hypoxia can also trigger mechanisms important for the maintenance of CSCs such as hypoxia-inducible factor (HIF) signaling, autophagy, and epithelial–mesenchymal transition (EMT) which is associated with acquisition of the mesenchymal characteristics by tumor cells. Recent data suggest that HIF signaling is important for the regulation of CSC properties in glioblastoma, ovarian cancer, and breast cancer, and also contributes to the activation of autophagy and EMT [49][52][53][54][55]. Autophagy is a mechanism of cellular “self-eating” by which cells sequestrate and digest certain organelles and protein complexes in a process of clean-up of damaged structures and misfolded proteins [56]. Autophagy might decrease efficiency of radiotherapy by its contribution to CSC maintenance and reduction of ROS-associated DNA damage [56][57][58]. A growing body of pre-clinical data demonstrated that EMT-related signaling pathways contribute to tumor cell reprogramming into CSCs, metastatic tumor spread, and radioresistance [59][60][61][62]. Taken together, these findings suggest that increasing the content of molecular oxygen in the tumor, or tumor reoxygenation, might be an effective way to increase radiation-induced DNA damage and to inhibit the signaling pathways and mechanisms favorable for CSC survival.
Tumor recurrences arise after radiotherapy depending on the actively proliferating tumor progenitor cells induced by tumor reoxygenation [63]. Tumor cell repopulation in the course of treatment might cause therapy failure. In support of this, clinical studies demonstrated that shortening the overall treatment time for fast-proliferating tumors prevents tumor repopulation and contributes to improved local tumor control [64]. The unlimited self-renewal capacity of CSCs suggests their role in tumor repopulation between the radiotherapy fractions. The recent computational modeling of HNSCC growth demonstrated that tumor re-oxygenation during treatment can result in substantial increase in the probability of CSC symmetric division (up to 50% as compared to 2% before treatment) that yields an increase of the CSC population within the tumor up to 30–35% [65].
Hereby, the radiobiological concept of 5Rs has to be considered in context of the cancer stem cell tumor model to explain the effect of radiotherapy on tumor tissues. In the next chapters, we discuss the DNA repair and further mechanisms of CSC therapy resistance to identify them as potential targets for the specific elimination of CSC populations.
3. Molecular Mechanisms of CSC Radioresistance
3.1. DNA Repair Factors and Pathways Upregulated in CSCs
It was previously observed that CSCs, comparable to tissue stem cells, have altered DNA damage response and repair pathways (Figure 2). In tissue stem cells, the alteration of DNA repair pathways ensures error-free DNA preservation. In CSCs, on the other hand, this causes resistance to exogenously induced DNA damage that leads to the failure of tumor therapy [66][67]. After irradiation, resistance correlates with significantly less DNA damage in glioma and breast cancer stem cells [68][69][70]. This also confirms the generally accepted view that tissue stem cells guarantee the maintenance of genomic stability and CSCs ensure the survival of the entire tumor population.
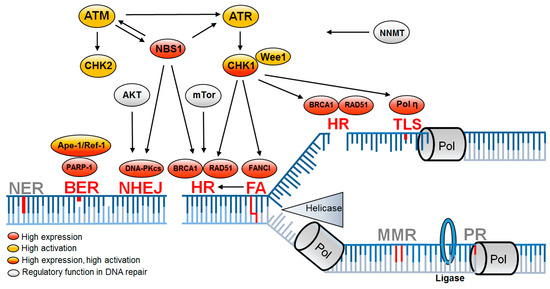
Figure 2. DNA repair pathways altered in CSCs are associated with DNA replication. Replication-associated DNA repair pathways leading to enhanced DNA repair (red) mediate resistance in CSCs, while further replication-associated DNA repair pathways remain unchanged (gray). Upregulation is based on increased expression (red) or increased activation (yellow) or both (red/yellow) and is supported by regulatory proteins (gray). BER—base excision repair; FA—Fanconi anemia; HR—homologous recombination; NER—nucleotide excision repair; NHEJ—non-homologous end joining; MMR—mismatch repair; PR—proof reading; TLS—translesion synthesis [9][68][71][72][73][74][75][76][77][78][79][80][81][82][83][84][85].
Most of the investigations regarding radioresistance and DNA repair were performed in glioblastoma. Radioresistance is mediated by enhanced activation of the two serine threonine protein kinases ataxia–telangiectasia mutated (ATM) and ATM- and Rad3-Related (ATR) and their two downstream checkpoint kinases (Chk1 and Chk2) [68][69][82][86][87]. Both kinases regulate the complex DNA damage response by initiating cell-cycle checkpoint control and activating corresponding DNA repair pathways. CD133+ glioma stem cells show an increased Chk1-dependent checkpoint response [9][69][86]. An increased expression of NBS1, a component of the Mre11, Rad50, Nbs1 (MRN) complex [88] involved in DSB sensor activity, was also detected. Increased DNA damage response and repair gene expression were also observed in CD133+ CSCs in lung carcinoma cells, breast, breast carcinoma-inducing cells, pancreatic CSCs, and non-small-cell lung cancer [41][71].
Starting point for the enhanced DNA damage response appears to be a general adaptation to increased replication stress and increased oxidative damage already in the untreated state, which leads to increased activation of the DNA damage response even after irradiation [84][89]. The DNA damage response is then mediated either by induced DSBs for amplified activation of ATM or by increased replicative stress for amplified activation of ATR. This results in a lower number of DSBs [89][90].
With regard to DNA double-strand break repair pathways in CSCs, HR is of outstanding importance, whereas, for NHEJ, only ATM-mediated effects are observed [91][92][93]. HR is the preferred DSB repair pathway in the S phase and it is more strongly activated by ATR and ATM in cells tolerating replication stress [68][72]. RAD51 overexpression, the main HR DNA repair protein, is observed in glioma stem cells and decreases at the transition to progenitor cells [92]. Glioblastoma cell lines show an upregulation of HR genes, especially RAD51, BRCA1, and BRCA2, which leads to less DNA damage after irradiation [74]. Thus, most studies suggest that resistance of CSCs manifests during the S phase by HR, regulated by MRN and activated by ATM/ATR.
The importance of S-phase repair also appears for CSC resistance mechanisms after chemotherapy in ovarian cancer. Here, resistance is observed by enhanced activation of translesion synthesis mediated by polymerase eta [79], as well as in HNSCC, where overexpression of the Fanconi anemia DNA repair proteins was observed in ALDH1-positive tumor cells [81].
However, a number of studies showed no difference or even lower DNA damage response in CSCs [41][91][94][95][96]. These contradictory observations suggest that an improved DNA damage response may not be a common feature of CSCs. More obvious is that CSCs and non-CSCs are transient populations and that, in addition to inter-tumoral heterogeneity, intratumoral heterogeneity must also be considered in DNA damage reaction functionality [96]. This was previously observed in glioblastoma, where RAD51 expression decreases from CSC to progenitor cells [92].
3.2. Factors Indirectly Influencing DNA Repair Capacity of CSCs
In CSCs, radioresistance determined by DNA repair capacity may also be attributed to lower indirectly induced ROS causing DNA damage and ROS-dependent apoptosis [14][70]. Breast CSCs express higher concentrations of ROS scavengers and neutralize radiation-induced ROS [89]. In addition to the known proteins with ROS scavenger function, the multifunctional protein apurine/apirimidine endonuclease/redox effector factor (Ape1/Ref-1) is also increasingly expressed in CSCs. Among other functions, Ape1/Ref-1 is part of the DNA repair complex base excision repair (BER), so that Ape1/Ref-1 can reduce both intracellular ROS and increase DNA repair [68]. Radioresistance in mesenchymal CSCs indirectly influencing DNA repair capacity could also be due to nicotinamide N-methyltransferase (NNMT) overexpression through depletion of the accessible amounts of nicotinamide, which is a known inhibitor of cellular DNA repair mechanisms [85].
An additional resistance mechanism only indirectly linked to improved DNA repair is the induction of CSC quiescence. By stopping the cells in the G0 phase, DNA damage can be eliminated before entering the S phase [44][97]. This shift in the cell cycle distribution may explain why NHEJ was considered important for increased radiation resistance in recent studies [68][76][98][99][100].
Another observed phenomenon indirectly linked to DNA repair mediated resistance is that genotoxic treatment, such as irradiation and chemotherapy, itself leads to the reprogramming of non-CSCs into CSCs [22][75][83][101][102][103][104][105]. In reprogrammed stem-like cells, either a faster repair mediated by BRCA1 and RAD51 after gemcitabine in pancreatic cancer [75] or a stronger activation of ATR/Chk1 in colon carcinoma after treatment with DNA interstrand-crosslinking (ICL) agents was shown [83]. Zhang and colleagues even went so far as to postulate a direct dependence of the DNA signaling cascade and stem-cell characteristics. They observed an ATM-mediated stabilization of zinc finger E-box binding homeobox 1 (ZEB1) leading to an enhanced Chk1-dependent DNA damage response in previously epithelial breast cells [104]. This direct dependence on stem cell character and HR or S-phase DNA repair was also observed for breast epithelial cells. Depletion of BRCA1 and FANCD2 led to reprogramming in breast epithelial cells to mesenchymal phenotype [105].
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386.
- McGuire, S. World Cancer Report 2014. Geneva, Switzerland: World Health Organization, International Agency for Research on Cancer, WHO Press, 2015. Adv. Nutr. 2016, 7, 418–419.
- Bray, F.; Jemal, A.; Grey, N.; Ferlay, J.; Forman, D. Global cancer transitions according to the Human Development Index (2008–2030): A population-based study. Lancet Oncol. 2012, 13, 790–801.
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Dicker, D.; Pain, A.; Hamavid, H.; Moradi-Lakeh, M.; MacIntyre, M.F.; Allen, C.; Hansen, G.; Woodbrook, R.; et al. The Global Burden of Cancer 2013. JAMA Oncol. 2015, 1, 505–527.
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2018.
- Atun, R.; Jaffray, D.A.; Barton, M.B.; Bray, F.; Baumann, M.; Vikram, B.; Hanna, T.P.; Knaul, F.M.; Lievens, Y.; Lui, T.Y.; et al. Expanding global access to radiotherapy. Lancet Oncol. 2015, 16, 1153–1186.
- Baumann, M.; Krause, M.; Overgaard, J.; Debus, J.; Bentzen, S.M.; Daartz, J.; Richter, C.; Zips, D.; Bortfeld, T. Radiation oncology in the era of precision medicine. Nat. Rev. Cancer 2016, 16, 234–249.
- Domina, E.A.; Philchenkov, A.; Dubrovska, A. Individual Response to Ionizing Radiation and Personalized Radiotherapy. Crit. Rev. Oncog. 2018, 23, 69–92.
- Baumann, M.; Krause, M.; Hill, R. Exploring the role of cancer stem cells in radioresistance. Nat. Rev. Cancer 2008, 8, 545–554.
- Butof, R.; Dubrovska, A.; Baumann, M. Clinical perspectives of cancer stem cell research in radiation oncology. Radiother. Oncol. 2013, 108, 388–396.
- Krause, M.; Dubrovska, A.; Linge, A.; Baumann, M. Cancer stem cells: Radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv. Drug Deliv. Rev. 2017, 109, 63–73.
- Krause, M.; Yaromina, A.; Eicheler, W.; Koch, U.; Baumann, M. Cancer stem cells: Targets and potential biomarkers for radiotherapy. Clin. Cancer Res. 2011, 17, 7224–7229.
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291.
- Peitzsch, C.; Kurth, I.; Kunz-Schughart, L.; Baumann, M.; Dubrovska, A. Discovery of the cancer stem cell related determinants of radioresistance. Radiother. Oncol. 2013, 108, 378–387.
- Peitzsch, C.; Tyutyunnykova, A.; Pantel, K.; Dubrovska, A. Cancer stem cells: The root of tumor recurrence and metastases. Semin. Cancer Biol. 2017.
- Hill, R.P.; Milas, L. The proportion of stem cells in murine tumors. Int. J. Radiat. Oncol. Biol. Phys. 1989, 16, 513–518.
- Baumann, M.; Dubois, W.; Suit, H.D. Response of human squamous cell carcinoma xenografts of different sizes to irradiation: Relationship of clonogenic cells, cellular radiation sensitivity in vivo, and tumor rescuing units. Radiat. Res. 1990, 123, 325–330.
- Koch, U.; Krause, M.; Baumann, M. Cancer stem cells at the crossroads of current cancer therapy failures--radiation oncology perspective. Semin. Cancer Biol. 2010, 20, 116–124.
- Dubben, H.H.; Thames, H.D.; Beck-Bornholdt, H.P. Tumor volume: A basic and specific response predictor in radiotherapy. Radiother. Oncol. 1998, 47, 167–174.
- Linge, A.; Dubrovska, A.; Baumann, M.; Krause, M. The role of cancer stem cells in tumour radioresponse. In Strategies to Enhance the Therapeutic Ratio of Radiation as a Cancer Treatment; Anscher, M., Valerie, K., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 43–74.
- Baccelli, I.; Trumpp, A. The evolving concept of cancer and metastasis stem cells. J. Cell Biol. 2012, 198, 281–293.
- Peitzsch, C.; Cojoc, M.; Hein, L.; Kurth, I.; Mabert, K.; Trautmann, F.; Klink, B.; Schrock, E.; Wirth, M.P.; Krause, M.; et al. An Epigenetic Reprogramming Strategy to Resensitize Radioresistant Prostate Cancer Cells. Cancer Res. 2016, 76, 2637–2651.
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134.
- Bedard, P.L.; Hansen, A.R.; Ratain, M.J.; Siu, L.L. Tumour heterogeneity in the clinic. Nature 2013, 501, 355–364.
- Razzouk, S. Translational genomics and head and neck cancer: Toward precision medicine. Clin. Genet. 2014, 86, 412–421.
- Steel, G.G.; McMillan, T.J.; Peacock, J.H. The 5Rs of radiobiology. Int. J. Radiat. Biol. 1989, 56, 1045–1048.
- Withers, H.R. The four R’s of radiotherapy. In Advances in Radiation Biology; Lett, J.T., Adler, H., Eds.; Academic Press: New York, NY, USA, 1975; Volume 5, pp. 241–271.
- Morgan, M.A.; Lawrence, T.S. Molecular Pathways: Overcoming Radiation Resistance by Targeting DNA Damage Response Pathways. Clin. Cancer Res. 2015, 21, 2898–2904.
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33.
- Pilie, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104.
- Azzam, E.I.; Jay-Gerin, J.P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60.
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462.
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591.
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA double-strand-break repair in higher eukaryotes and its role in genomic instability and cancer: Cell cycle and proliferation-dependent regulation. Semin. Cancer Biol. 2016, 37–38, 51–64.
- Hufnagl, A.; Herr, L.; Friedrich, T.; Durante, M.; Taucher-Scholz, G.; Scholz, M. The link between cell-cycle dependent radiosensitivity and repair pathways: A model based on the local, sister-chromatid conformation dependent switch between NHEJ and HR. Dna. Repair 2015, 27, 28–39.
- Lieber, M.R.; Ma, Y.; Pannicke, U.; Schwarz, K. Mechanism and regulation of human non-homologous DNA end-joining. Nat. Rev. Mol. Cell Biol. 2003, 4, 712–720.
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902–2906.
- Mjelle, R.; Hegre, S.A.; Aas, P.A.; Slupphaug, G.; Drablos, F.; Saetrom, P.; Krokan, H.E. Cell cycle regulation of human DNA repair and chromatin remodeling genes. Dna. Repair 2015, 30, 53–67.
- Kakarougkas, A.; Jeggo, P.A. DNA DSB repair pathway choice: An orchestrated handover mechanism. Br. J. Radiol. 2014, 87, 20130685.
- Karanam, K.; Kafri, R.; Loewer, A.; Lahav, G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol. Cell 2012, 47, 320–329.
- Borgmann, K.; Kocher, S.; Kriegs, M.; Mansour, W.Y.; Parplys, A.C.; Rieckmann, T.; Rothkamm, K. DNA Repair. In Molecular Radio-Oncology, Recent Results in Cancer Research; Springer: Berlin/Heidelberg, Germany, 2016; Volume 198, pp. 1–24.
- Moore, N.; Lyle, S. Quiescent, slow-cycling stem cell populations in cancer: A review of the evidence and discussion of significance. J. Oncol. 2011, 2011, 396076.
- Ambrosio, S.; Di Palo, G.; Napolitano, G.; Amente, S.; Dellino, G.I.; Faretta, M.; Pelicci, P.G.; Lania, L.; Majello, B. Cell cycle-dependent resolution of DNA double-strand breaks. Oncotarget 2016, 7, 4949–4960.
- Chen, Y.; Li, D.; Wang, D.; Liu, X.; Yin, N.; Song, Y.; Lu, S.H.; Ju, Z.; Zhan, Q. Quiescence and attenuated DNA damage response promote survival of esophageal cancer stem cells. J. Cell. Biochem. 2012, 113, 3643–3652.
- Chang, L.; Graham, P.; Hao, J.; Ni, J.; Deng, J.; Bucci, J.; Malouf, D.; Gillatt, D.; Li, Y. Cancer stem cells and signaling pathways in radioresistance. Oncotarget 2016, 7, 11002–11017.
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828.
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833.
- Hockel, M.; Vaupel, P. Tumor hypoxia: Definitions and current clinical, biologic, and molecular aspects. J. Natl. Cancer Inst. 2001, 93, 266–276.
- Peitzsch, C.; Perrin, R.; Hill, R.P.; Dubrovska, A.; Kurth, I. Hypoxia as a biomarker for radioresistant cancer stem cells. Int. J. Radiat. Biol. 2014, 90, 636–652.
- Gallamini, A.; Zwarthoed, C.; Borra, A. Positron Emission Tomography (PET) in Oncology. Cancers 2014, 6, 1821–1889.
- Abramyuk, A.; Appold, S.; Zophel, K.; Baumann, M.; Abolmaali, N. Modification of staging and treatment of head and neck cancer by FDG-PET/CT prior to radiotherapy. Strahlenther. Onkol. 2013, 189, 197–201.
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238.
- Wang, P.; Lan, C.; Xiong, S.; Zhao, X.; Shan, Y.; Hu, R.; Wan, W.; Yu, S.; Liao, B.; Li, G.; et al. HIF1alpha regulates single differentiated glioma cell dedifferentiation to stem-like cell phenotypes with high tumorigenic potential under hypoxia. Oncotarget 2017, 8, 28074–28092.
- Luo, M.; Wicha, M.S. Targeting Cancer Stem Cell Redox Metabolism to Enhance Therapy Responses. Semin. Radiat. Oncol. 2019, 29, 42–54.
- Qin, J.; Liu, Y.; Lu, Y.; Liu, M.; Li, M.; Li, J.; Wu, L. Hypoxia-inducible factor 1 alpha promotes cancer stem cells-like properties in human ovarian cancer cells by upregulating SIRT1 expression. Sci. Rep. 2017, 7, 10592.
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22.
- Feng, H.; Wang, J.; Chen, W.; Shan, B.; Guo, Y.; Xu, J.; Wang, L.; Guo, P.; Zhang, Y. Hypoxia-induced autophagy as an additional mechanism in human osteosarcoma radioresistance. J. Bone Oncol. 2016, 5, 67–73.
- Hou, J.; Han, Z.P.; Jing, Y.Y.; Yang, X.; Zhang, S.S.; Sun, K.; Hao, C.; Meng, Y.; Yu, F.H.; Liu, X.Q.; et al. Autophagy prevents irradiation injury and maintains stemness through decreasing ROS generation in mesenchymal stem cells. Cell Death Dis. 2013, 4, e844.
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629.
- de Jong, M.C.; Ten Hoeve, J.J.; Grenman, R.; Wessels, L.F.; Kerkhoven, R.; Te Riele, H.; van den Brekel, M.W.; Verheij, M.; Begg, A.C. Pretreatment microRNA Expression Impacting on Epithelial-to-Mesenchymal Transition Predicts Intrinsic Radiosensitivity in Head and Neck Cancer Cell Lines and Patients. Clin. Cancer Res. 2015, 21, 5630–5638.
- Huang, T.; Kim, C.K.; Alvarez, A.A.; Pangeni, R.P.; Wan, X.; Song, X.; Shi, T.; Yang, Y.; Sastry, N.; Horbinski, C.M.; et al. MST4 Phosphorylation of ATG4B Regulates Autophagic Activity, Tumorigenicity, and Radioresistance in Glioblastoma. Cancer Cell 2017, 32, 840–855.
- Huang, T.; Wan, X.; Alvarez, A.A.; James, C.D.; Song, X.; Yang, Y.; Sastry, N.; Nakano, I.; Sulman, E.P.; Hu, B.; et al. MIR93 (microRNA-93) Regulates Tumorigenicity and Therapy Response of Glioblastoma by Targeting Autophagy. Autophagy 2019.
- Chan, R.; Sethi, P.; Jyoti, A.; McGarry, R.; Upreti, M. Investigating the Radioresistant Properties of Lung Cancer Stem Cells in the Context of the Tumor Microenvironment. Radiat. Res. 2016, 185, 169–181.
- Butof, R.; Baumann, M. Time in radiation oncology—Keep it short! Radiother. Oncol. 2013, 106, 271–275.
- Wu, L.; Blum, W.; Zhu, C.Q.; Yun, Z.; Pecze, L.; Kohno, M.; Chan, M.L.; Zhao, Y.; Felley-Bosco, E.; Schwaller, B.; et al. Putative cancer stem cells may be the key target to inhibit cancer cell repopulation between the intervals of chemoradiation in murine mesothelioma. BMC Cancer 2018, 18, 471.
- Maynard, S.; Schurman, S.H.; Harboe, C.; de Souza-Pinto, N.C.; Bohr, V.A. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis 2009, 30, 2–10.
- Mandal, P.K.; Blanpain, C.; Rossi, D.J. DNA damage response in adult stem cells: Pathways and consequences. Nat. Rev. Mol. Cell Biol. 2011, 12, 198–202.
- Vitale, I.; Manic, G.; De Maria, R.; Kroemer, G.; Galluzzi, L. DNA Damage in Stem Cells. Mol. Cell 2017, 66, 306–319.
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760.
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J. Natl. Cancer Inst. 2006, 98, 1777–1785.
- Desai, A.; Webb, B.; Gerson, S.L. CD133+ cells contribute to radioresistance via altered regulation of DNA repair genes in human lung cancer cells. Radiother. Oncol. 2014, 110, 538–545.
- Lim, Y.C.; Roberts, T.L.; Day, B.W.; Harding, A.; Kozlov, S.; Kijas, A.W.; Ensbey, K.S.; Walker, D.G.; Lavin, M.F. A role for homologous recombination and abnormal cell-cycle progression in radioresistance of glioma-initiating cells. Mol. Cancer Ther. 2012, 11, 1863–1872.
- Liu, Y.; Burness, M.L.; Martin-Trevino, R.; Guy, J.; Bai, S.; Harouaka, R.; Brooks, M.D.; Shang, L.; Fox, A.; Luther, T.K.; et al. RAD51 Mediates Resistance of Cancer Stem Cells to PARP Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 514–522.
- Balbous, A.; Cortes, U.; Guilloteau, K.; Rivet, P.; Pinel, B.; Duchesne, M.; Godet, J.; Boissonnade, O.; Wager, M.; Bensadoun, R.J.; et al. A radiosensitizing effect of RAD51 inhibition in glioblastoma stem-like cells. BMC Cancer 2016, 16, 604.
- Mathews, L.A.; Cabarcas, S.M.; Hurt, E.M.; Zhang, X.; Jaffee, E.M.; Farrar, W.L. Increased expression of DNA repair genes in invasive human pancreatic cancer cells. Pancreas 2011, 40, 730–739.
- Wang, Y.; Xu, H.; Liu, T.; Huang, M.; Butter, P.P.; Li, C.; Zhang, L.; Kao, G.D.; Gong, Y.; Maity, A.; et al. Temporal DNA-PK activation drives genomic instability and therapy resistance in glioma stem cells. JCI Insight 2018, 3, 98096.
- Karimi-Busheri, F.; Rasouli-Nia, A.; Mackey, J.R.; Weinfeld, M. Senescence evasion by MCF-7 human breast tumor-initiating cells. Breast Cancer Res. 2010, 12, R31.
- Gilabert, M.; Launay, S.; Ginestier, C.; Bertucci, F.; Audebert, S.; Pophillat, M.; Toiron, Y.; Baudelet, E.; Finetti, P.; Noguchi, T.; et al. Poly(ADP-ribose) polymerase 1 (PARP1) overexpression in human breast cancer stem cells and resistance to olaparib. PLoS ONE 2014, 9, e104302.
- Srivastava, A.K.; Han, C.; Zhao, R.; Cui, T.; Dai, Y.; Mao, C.; Zhao, W.; Zhang, X.; Yu, J.; Wang, Q.E. Enhanced expression of DNA polymerase eta contributes to cisplatin resistance of ovarian cancer stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, 4411–4416.
- Mir, S.E.; De Witt Hamer, P.C.; Krawczyk, P.M.; Balaj, L.; Claes, A.; Niers, J.M.; Van Tilborg, A.A.; Zwinderman, A.H.; Geerts, D.; Kaspers, G.J.; et al. In silico analysis of kinase expression identifies WEE1 as a gatekeeper against mitotic catastrophe in glioblastoma. Cancer Cell 2010, 18, 244–257.
- Wu, J.; Mu, Q.; Thiviyanathan, V.; Annapragada, A.; Vigneswaran, N. Cancer stem cells are enriched in Fanconi anemia head and neck squamous cell carcinomas. Int. J. Oncol. 2014, 45, 2365–2372.
- Ahmed, S.U.; Carruthers, R.; Gilmour, L.; Yildirim, S.; Watts, C.; Chalmers, A.J. Selective Inhibition of Parallel DNA Damage Response Pathways Optimizes Radiosensitization of Glioblastoma Stem-like Cells. Cancer Res. 2015, 75, 4416–4428.
- Gallmeier, E.; Hermann, P.C.; Mueller, M.T.; Machado, J.G.; Ziesch, A.; De Toni, E.N.; Palagyi, A.; Eisen, C.; Ellwart, J.W.; Rivera, J.; et al. Inhibition of ataxia telangiectasia- and Rad3-related function abrogates the in vitro and in vivo tumorigenicity of human colon cancer cells through depletion of the CD133(+) tumor-initiating cell fraction. Stem Cells 2011, 29, 418–429.
- Carruthers, R.D.; Ahmed, S.U.; Ramachandran, S.; Strathdee, K.; Kurian, K.M.; Hedley, A.; Gomez-Roman, N.; Kalna, G.; Neilson, M.; Gilmour, L.; et al. Replication Stress Drives Constitutive Activation of the DNA Damage Response and Radioresistance in Glioblastoma Stem-like Cells. Cancer Res. 2018, 78, 5060–5071.
- D’Andrea, F.P.; Safwat, A.; Kassem, M.; Gautier, L.; Overgaard, J.; Horsman, M.R. Cancer stem cell overexpression of nicotinamide N-methyltransferase enhances cellular radiation resistance. Radiother. Oncol. 2011, 99, 373–378.
- Bartucci, M.; Svensson, S.; Romania, P.; Dattilo, R.; Patrizii, M.; Signore, M.; Navarra, S.; Lotti, F.; Biffoni, M.; Pilozzi, E.; et al. Therapeutic targeting of Chk1 in NSCLC stem cells during chemotherapy. Cell Death Differ. 2012, 19, 768–778.
- Carruthers, R.; Ahmed, S.U.; Strathdee, K.; Gomez-Roman, N.; Amoah-Buahin, E.; Watts, C.; Chalmers, A.J. Abrogation of radioresistance in glioblastoma stem-like cells by inhibition of ATM kinase. Mol. Oncol. 2015, 9, 192–203.
- Cheng, L.; Wu, Q.; Huang, Z.; Guryanova, O.A.; Huang, Q.; Shou, W.; Rich, J.N.; Bao, S. L1CAM regulates DNA damage checkpoint response of glioblastoma stem cells through NBS1. EMBO J. 2011, 30, 800–813.
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783.
- Bartkova, J.; Hamerlik, P.; Stockhausen, M.T.; Ehrmann, J.; Hlobilkova, A.; Laursen, H.; Kalita, O.; Kolar, Z.; Poulsen, H.S.; Broholm, H.; et al. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene 2010, 29, 5095–5102.
- Lundholm, L.; Haag, P.; Zong, D.; Juntti, T.; Mork, B.; Lewensohn, R.; Viktorsson, K. Resistance to DNA-damaging treatment in non-small cell lung cancer tumor-initiating cells involves reduced DNA-PK/ATM activation and diminished cell cycle arrest. Cell Death Dis. 2013, 4, e478.
- King, H.O.; Brend, T.; Payne, H.L.; Wright, A.; Ward, T.A.; Patel, K.; Egnuni, T.; Stead, L.F.; Patel, A.; Wurdak, H.; et al. RAD51 Is a Selective DNA Repair Target to Radiosensitize Glioma Stem Cells. Stem Cell Rep. 2017, 8, 125–139.
- Ying, S.; Chen, Z.; Medhurst, A.L.; Neal, J.A.; Bao, Z.; Mortusewicz, O.; McGouran, J.; Song, X.; Shen, H.; Hamdy, F.C.; et al. DNA-PKcs and PARP1 Bind to Unresected Stalled DNA Replication Forks Where They Recruit XRCC1 to Mediate Repair. Cancer Res. 2016, 76, 1078–1088.
- McCord, A.M.; Jamal, M.; Williams, E.S.; Camphausen, K.; Tofilon, P.J. CD133+ glioblastoma stem-like cells are radiosensitive with a defective DNA damage response compared with established cell lines. Clin. Cancer Res. 2009, 15, 5145–5153.
- Ropolo, M.; Daga, A.; Griffero, F.; Foresta, M.; Casartelli, G.; Zunino, A.; Poggi, A.; Cappelli, E.; Zona, G.; Spaziante, R.; et al. Comparative analysis of DNA repair in stem and nonstem glioma cell cultures. Mol. Cancer Res. 2009, 7, 383–392.
- Magee, J.A.; Piskounova, E.; Morrison, S.J. Cancer stem cells: Impact, heterogeneity, and uncertainty. Cancer Cell 2012, 21, 283–296.
- Dembinski, J.L.; Krauss, S. Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin. Exp. Metastasis 2009, 26, 611–623.
- Facchino, S.; Abdouh, M.; Chatoo, W.; Bernier, G. BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery. J. Neurosci. 2010, 30, 10096–10111.
- Timme, C.R.; Rath, B.H.; O’Neill, J.W.; Camphausen, K.; Tofilon, P.J. The DNA-PK Inhibitor VX-984 Enhances the Radiosensitivity of Glioblastoma Cells Grown In Vitro and as Orthotopic Xenografts. Mol. Cancer Ther. 2018, 17, 1207–1216.
- Maugeri-Sacca, M.; Bartucci, M.; De Maria, R. DNA damage repair pathways in cancer stem cells. Mol. Cancer Ther. 2012, 11, 1627–1636.
- Ghisolfi, L.; Keates, A.C.; Hu, X.; Lee, D.K.; Li, C.J. Ionizing radiation induces stemness in cancer cells. PLoS ONE 2012, 7, e43628.
- Lagadec, C.; Vlashi, E.; Della Donna, L.; Dekmezian, C.; Pajonk, F. Radiation-induced reprogramming of breast cancer cells. Stem Cells 2012, 30, 833–844.
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1787.
- Zhang, P.; Wei, Y.; Wang, L.; Debeb, B.G.; Yuan, Y.; Zhang, J.; Yuan, J.; Wang, M.; Chen, D.; Sun, Y.; et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat. Cell Biol. 2014, 16, 864–875.
- Wang, H.; Bierie, B.; Li, A.G.; Pathania, S.; Toomire, K.; Dimitrov, S.D.; Liu, B.; Gelman, R.; Giobbie-Hurder, A.; Feunteun, J.; et al. BRCA1/FANCD2/BRG1-Driven DNA Repair Stabilizes the Differentiation State of Human Mammary Epithelial Cells. Mol. Cell 2016, 63, 277–292.
More
Information
Subjects:
Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
900
Revisions:
2 times
(View History)
Update Date:
08 Sep 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No