 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kelly Johanna Hidalgo Martinez | + 5224 word(s) | 5224 | 2021-09-03 06:23:14 |
Video Upload Options
Microorganisms inhabiting subsurface petroleum reservoirs are key players in biochemical transformations. The interactions of microbial communities in these environments are highly complex and still poorly understood. This work aimed to assess publicly available metagenomes from oil reservoirs and implement a robust pipeline of genome-resolved metagenomics to decipher metabolic and taxonomic profiles of petroleum reservoirs worldwide. We noticed that the oil reservoirs with a lower level of intervention were the most similar to the potential functional core, while the oil fields with a long history of water injection had greater variation in functional profile. These results show how key microorganisms and their functions respond to the distinct physicochemical parameters and interventions of the oil field operations such as water injection and expand the knowledge of biogeochemical transformations in these ecosystems.
1. Introduction
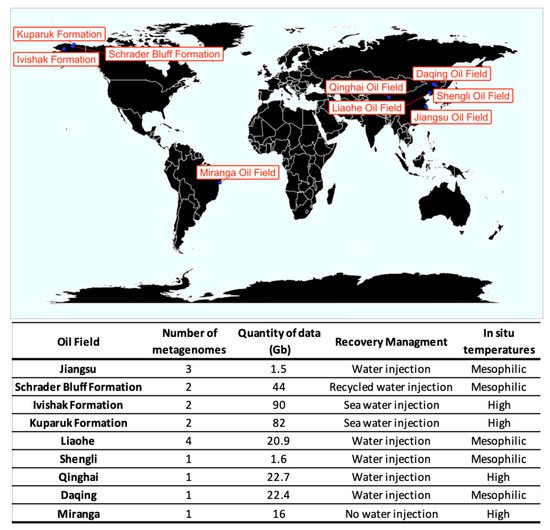
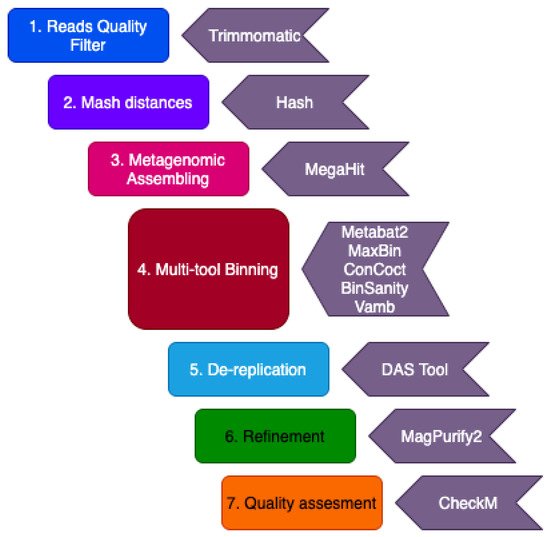
2. Description of Datasets
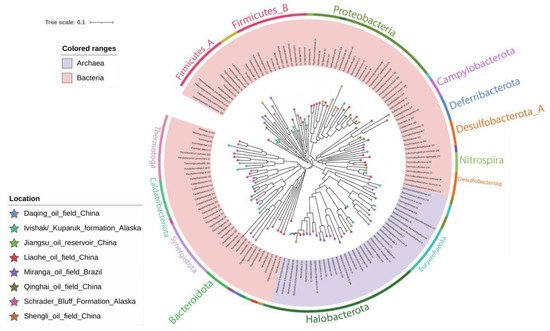
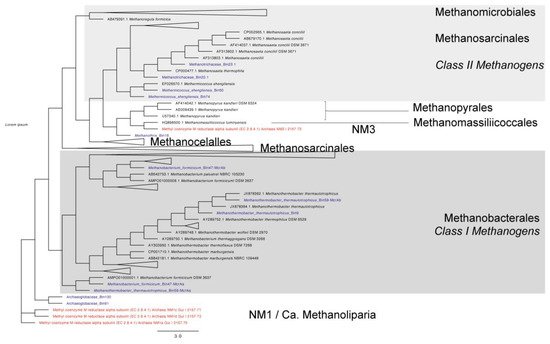
3. Taxonomic Affiliation and Phylogeny
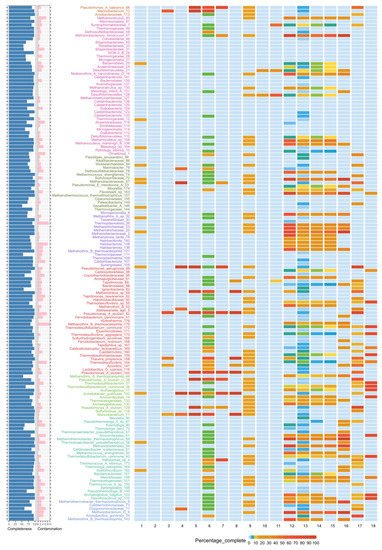
3.4. Metabolic Potential
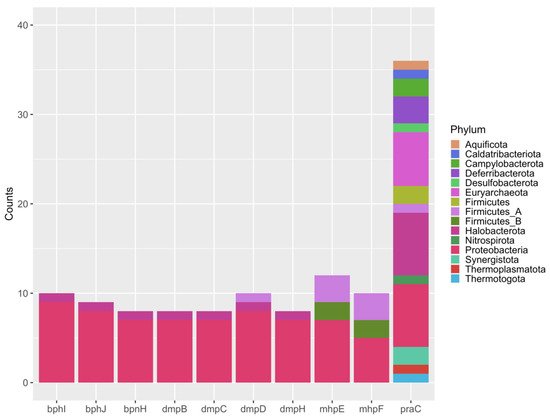
3.5. Functional Core Analysis
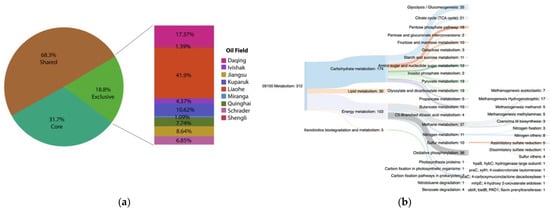
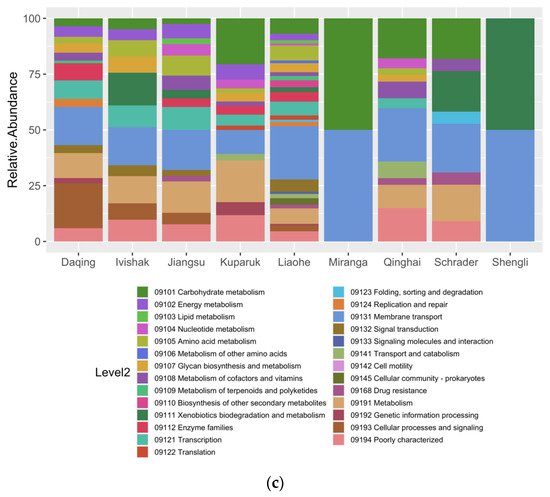
Environment-Specific Orthogroups
4. Conclusions
References
- Wang, X.; Li, X.; Yu, L.; Huang, L.; Xiu, J.; Lin, W.; Zhang, Y. Characterizing the microbiome in petroleum reservoir flooded by different water sources. Sci. Total Environ. 2018, 653, 872–885.
- Nie, Y.; Zhao, J.-Y.; Tang, Y.-Q.; Guo, P.; Yang, Y.; Wu, X.-L.; Zhao, F. Species divergence vs. functional convergence characterizes crude oil microbial community assembly. Front. Microbiol. 2016, 7, 1254.
- Sierra-Garcia, I.N.; Belgini, D.R.B.; Torres-Ballesteros, A.; Paez-Espino, D.; Capilla, R.; Santos Neto, E.V.; Gray, N.; de Oliveira, V.M. In depth metagenomic analysis in contrasting oil wells reveals syntrophic bacterial and archaeal associations for oil biodegradation in petroleum reservoirs. Sci. Total Environ. 2020, 715, 136646.
- Sierra-Garcia, I.N.; Dellagnezze, B.M.; Santos, V.P.; Capilla, R.; Santos Neto, E.V.S.; Gray, N.; Oliveira, V.M. Microbial diversity in degraded and non-degraded petroleum samples and comparison across oil reservoirs at local and global scales. Extremophiles 2017, 21, 211–229.
- An, D.; Caffrey, S.M.; Soh, J.; Agrawal, A.; Brown, D.; Budwill, K.; Dong, X.; Dunfield, P.F.; Foght, J.; Gieg, L.M.; et al. Metagenomics of hydrocarbon resource environments indicates aerobic taxa and genes to be unexpectedly common. Environ. Sci. Technol. 2013, 47, 10708–10717.
- Hu, P.; Tom, L.; Singh, A.; Thomas, B.C.; Baker, B.J.; Piceno, Y.M.; Andersen, G.L.; Banfield, J.F. Genome-resolved metagenomic analysis reveals roles for candidate phyla and other microbial community members in biogeochemical transformations in oil reservoirs. mBio 2016, 7, e01669-15.
- Kotlar, H.K.; Lewin, A.; Johansen, J.; Throne-Holst, M.; Haverkamp, T.; Markussen, S.; Winnberg, A.; Ringrose, P.; Aakvik, T.; Ryeng, E. High coverage sequencing of DNA from microorganisms living in an oil reservoir 2.5 kilometres subsurface. Environ. Microbiol. Rep. 2011, 3, 674–681.
- Hug, L.A.; Baker, B.J.; Anantharaman, K.; Brown, C.T.; Probst, A.J.; Castelle, C.J.; Butterfield, C.N.; Hernsdorf, A.W.; Amano, Y.; Ise, K.; et al. A new view of the tree of life. Nat. Microbiol. 2016, 1, 16048.
- Parks, D.H.; Rinke, C.; Chuvochina, M.; Chaumeil, P.-A.; Woodcroft, B.J.; Evans, P.N.; Hugenholtz, P.; Tyson, G.W. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2017, 2, 1533–1542.
- Tyson, G.W.; Chapman, J.; Hugenholtz, P.; Allen, E.E.; Ram, R.J.; Richardson, P.M.; Solovyev, V.V.; Rubin, E.M.; Rokhsar, D.S.; Banfield, J.F. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 2004, 428, 37–43.
- Liu, Y.-F.; Galzerani, D.D.; Mbadinga, S.M.; Zaramela, L.S.; Gu, J.-D.; Mu, B.-Z.; Zengler, K. Metabolic capability and in situ activity of microorganisms in an oil reservoir. Microbiome 2018, 6, 5.
- Song, Z.; Chen, S.; Zhao, F.; Zhu, W. Whole metagenome of injected and produced fluids reveal the heterogenetic characteristics of the microbial community in a water-flooded oil reservoir. J. Pet. Sci. Eng. 2019, 176, 1198–1207.
- Vigneron, A.; Alsop, E.B.; Lomans, B.P.; Kyrpides, N.C.; Head, I.M.; Tsesmetzis, N. Succession in the petroleum reservoir microbiome through an oil field production lifecycle. ISME J. 2017, 11, 2141–2154.
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.-A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004.
- Brown, C.T.; Hug, L.A.; Thomas, B.C.; Sharon, I.; Castelle, C.J.; Singh, A.; Wilkins, M.J.; Wrighton, K.C.; Williams, K.H.; Banfield, J.F. Unusual biology across a group comprising more than 15% of domain bacteria. Nature 2015, 523, 208–211.
- Frey, B.; Rime, T.; Phillips, M.; Stierli, B.; Hajdas, I.; Widmer, F.; Hartmann, M. Microbial diversity in European alpine permafrost and active layers. FEMS Microbiol. Ecol. 2016, 92, fiw018.
- Herrmann, M.; Wegner, C.-E.; Taubert, M.; Geesink, P.; Lehmann, K.; Yan, L.; Lehmann, R.; Totsche, K.U.; Küsel, K. Predominance of cand. patescibacteria in groundwater is caused by their preferential mobilization from soils and flourishing under oligotrophic conditions. Front. Microbiol. 2019, 10, 1407.
- Hubalek, V.; Wu, X.; Eiler, A.; Buck, M.; Heim, C.; Dopson, M.; Bertilsson, S.; Ionescu, D. Connectivity to the surface determines diversity patterns in subsurface aquifers of the Fennoscandian shield. ISME J. 2016, 10, 2447–2458.
- León-Zayas, R.; Peoples, L.; Biddle, J.F.; Podell, S.; Novotny, M.; Cameron, J.; Lasken, R.S.; Bartlett, D.H. The metabolic potential of the single cell genomes obtained from the Challenger Deep, Mariana Trench within the candidate superphylum P arcubacteria (OD 1). Environ. Microbiol. 2017, 19, 2769–2784.
- Luef, B.; Frischkorn, K.R.; Wrighton, K.C.; Holman, H.-Y.N.; Birarda, G.; Thomas, B.C.; Singh, A.; Williams, K.H.; Siegerist, C.E.; Tringe, S.G.; et al. Diverse uncultivated ultra-small bacterial cells in groundwater. Nat. Commun. 2015, 6, 6372.
- Antunes, T.C.; Marconatto, L.; Borges, L.G.d.A.; Giongo, A.; Sand, S.T.V.D. Analysis of microbial community biodiversity in activated sludge from a petrochemical plant. Rev. Ambiente Água 2021, 16.
- Eze, M.O.; Lütgert, S.A.; Neubauer, H.; Balouri, A.; Kraft, A.A.; Sieven, A.; Daniel, R.; Wemheuer, B. Metagenome assembly and metagenome-assembled genome sequences from a historical oil field located in Wietze, Germany. Microbiol. Resour. Announc. 2020, 9.
- Tian, R.; Ning, D.; He, Z.; Zhang, P.; Spencer, S.J.; Gao, S.; Shi, W.; Wu, L.; Zhang, Y.; Yang, Y.; et al. Small and mighty: Adaptation of superphylum Patescibacteria to groundwater environment drives their genome simplicity. Microbiome 2020, 8, 51.
- Castelle, C.J.; Brown, C.T.; Anantharaman, K.; Probst, A.J.; Huang, R.H.; Banfield, J.F. Biosynthetic capacity, metabolic variety and unusual biology in the CPR and DPANN radiations. Nat. Rev. Microbiol. 2018, 16, 629–645.
- Shelton, J.L.; Andrews, R.S.; Akob, D.M.; DeVera, C.A.; Mumford, A.; McCray, J.E.; McIntosh, J.C. Microbial community composition of a hydrocarbon reservoir 40 years after a CO2 enhanced oil recovery flood. FEMS Microbiol. Ecol. 2018, 94.
- Alsouleman, K.; Linke, B.; Klang, J.; Klocke, M.; Krakat, N.; Theuerl, S. Reorganisation of a mesophilic biogas microbiome as response to a stepwise increase of ammonium nitrogen induced by poultry manure supply. Bioresour. Technol. 2016, 208, 200–204.
- Gao, P.; Tian, H.; Wang, Y.; Li, Y.; Li, Y.; Xie, J.; Zeng, B.; Zhou, J.; Li, G.; Ma, T. Spatial isolation and environmental factors drive distinct bacterial and archaeal communities in different types of petroleum reservoirs in China. Sci. Rep. 2016, 6, 20174.
- Zhao, J.-Y.; Hu, B.; Dolfing, J.; Li, Y.; Tang, Y.-Q.; Jiang, Y.; Chi, C.-Q.; Xing, J.; Nie, Y.; Wu, X.-L. Thermodynamically favorable reactions shape the archaeal community affecting bacterial community assembly in oil reservoirs. Sci. Total Environ. 2021, 781, 146506.
- Wang, H.-Z.; Gou, M.; Yi, Y.; Xia, Z.-Y.; Tang, Y.-Q. Identification of novel potential acetate-oxidizing bacteria in an acetate-fed methanogenic chemostat based on DNA stable isotope probing. J. Gen. Appl. Microbiol. 2018, 64, 221–231.
- Sengupta, K.; Pal, S. A review on microbial diversity and genetic markers involved in methanogenic degradation of hydrocarbons: Futuristic prospects of biofuel recovery from contaminated regions. Environ. Sci. Pollut. Res. 2021, 28, 40288–40307.
- Oren, A. The family Methanotrichaceae. In The Prokaryotes; Springer: Berlin/Heidelberg, Gemany, 2014; pp. 298–306.
- Akinyemi, T.S.; Shao, N.; Whitman, W.B. Methanothrix. In Bergey’s Manual of Systematics of Archaea and Bacteria; Springer: Berlin/Heidelberg, Germany, 2014; pp. 297–306.
- Holmes, D.E.; Shrestha, P.M.; Walker, D.J.F.; Dang, Y.; Nevin, K.P.; Woodard, T.L.; Lovley, D.R. Metatranscriptomic evidence for direct interspecies electron transfer between geobacter and methanothrix species in methanogenic rice paddy soils. Appl. Environ. Microbiol. 2017, 83, e00223-17.
- John, E.S.; Flores, G.E.; Meneghin, J.; Reysenbach, A.-L. Deep-sea hydrothermal vent metagenome–Assembled genomes provide insight into the phylum Nanoarchaeota. Environ. Microbiol. Rep. 2019, 11, 262–270.
- Munson-McGee, J.H.; Field, E.K.; Bateson, M.; Rooney, C.; Stepanauskas, R.; Young, M.J.; Wommack, K.E. Nanoarchaeota, their sulfolobales host, and nanoarchaeota virus distribution across Yellowstone national park hot springs. Appl. Environ. Microbiol. 2015, 81, 7860–7868.
- Carrier, V.; Svenning, M.M.; Gründger, F.; Niemann, H.; Dessandier, P.-A.; Panieri, G.; Kalenitchenko, D. The impact of methane on microbial communities at marine arctic gas hydrate bearing sediment. Front. Microbiol. 2020, 11, 1932.
- Casanueva, A.; Galada, N.; Baker, G.C.; Grant, W.D.; Heaphy, S.; Jones, B.; Yanhe, M.; Ventosa, A.; Blamey, J.; Cowan, D.A. Nanoarchaeal 16S rRNA gene sequences are widely dispersed in hyperthermophilic and mesophilic halophilic environments. Extremophiles 2008, 12, 651–656.
- Chernyh, N.A.; Mardanov, A.V.; Gumerov, V.M.; Miroshnichenko, M.L.; Lebedinsky, A.V.; Merkel, A.Y.; Crowe, D.; Pimenov, N.V.; Rusanov, I.I.; Ravin, N.V.; et al. Microbial life in Bourlyashchy, the hottest thermal pool of Uzon Caldera, Kamchatka. Extremophiles 2015, 19, 1157–1171.
- Clingenpeel, S.; Kan, J.; Macur, R.; Woyke, T.; Lovalvo, D.; Varley, J.; Inskeep, W.P.; Nealson, K.; McDermott, T. Yellowstone lake nanoarchaeota. Front. Microbiol. 2013, 4, 274.
- Flores, G.; Shakya, M.; Meneghin, J.; Yang, Z.; Seewald, J.; Geoff Wheat, C.; Podar, M.; Reysenbach, A.L. Inter-field variability in the microbial communities of hydrothermal vent deposits from a back-arc basin. Geobiology 2012, 10, 333–346.
- Flores, G.E.; Campbell, J.H.; Kirshtein, J.D.; Meneghin, J.; Podar, M.; Steinberg, J.I.; Seewald, J.S.; Tivey, M.K.; Voytek, M.A.; Yang, Z.K.; et al. Microbial community structure of hydrothermal deposits from geochemically different vent fields along the Mid-Atlantic Ridge. Environ. Microbiol. 2011, 13, 2158–2171.
- Hohn, M.J.; Hedlund, B.P.; Huber, H. Detection of 16S rDNA sequences representing the novel phylum “Nanoarchaeota”: Indication for a wide distribution in high temperature biotopes. Syst. Appl. Microbiol. 2002, 25, 551–554.
- McCliment, E.A.; Voglesonger, K.M.; O’Day, P.A.; Dunn, E.E.; Holloway, J.R.; Cary, S.C. Colonization of nascent, deep-sea hydrothermal vents by a novel Archaeal and Nanoarchaeal assemblage. Environ. Microbiol. 2006, 8, 114–125.
- Zemskaya, T.I.; Cabello-Yeves, P.J.; Pavlova, O.N.; Rodriguez-Valera, F. Microorganisms of lake Baikal—The deepest and most ancient lake on Earth. Appl. Microbiol. Biotechnol. 2020, 104, 6079–6090.
- Mayumi, D.; Mochimaru, H.; Tamaki, H.; Yamamoto, K.; Yoshioka, H.; Suzuki, Y.; Kamagata, Y.; Sakata, S. Methane production from coal by a single methanogen. Science 2016, 354, 222–225.
- Cheng, L.; Ding, C.; Li, Q.; He, Q.; Dai, L.-R.; Zhang, H. DNA-SIP reveals that syntrophaceae play an important role in methanogenic hexadecane degradation. PLoS ONE 2013, 8, e66784.
- Gao, P.; Tian, H.; Li, G.; Sun, H.; Ma, T. Microbial diversity and abundance in the Xinjiang Luliang long-term water-flooding petroleum reservoir. Microbiol. Open 2015, 4, 332–342.
- Walker, D.J.F.; Adhikari, R.Y.; Holmes, D.E.; Ward, J.E.; Woodard, T.L.; Nevin, K.P.; Lovley, D.R. Electrically conductive pili from pilin genes of phylogenetically diverse microorganisms. ISME J. 2018, 12, 48–58.
- Greene, A.C.; Patel, B.K.C.; Yacob, S. Geoalkalibacter subterraneus sp. nov., an anaerobic Fe(III)- and Mn(IV)-reducing bacterium from a petroleum reservoir, and emended descriptions of the family Desulfuromonadaceae and the genus Geoalkalibacter. Int. J. Syst. Evol. Microbiol. 2009, 59, 781–785.
- Piceno, Y.M.; Reid, F.C.; Tom, L.M.; Conrad, M.E.; Bill, M.; Hubbard, C.G.; Fouke, B.W.; Graff, C.J.; Han, J.; Stringfellow, W.T.; et al. Temperature and injection water source influence microbial community structure in four Alaskan North Slope hydrocarbon reservoirs. Front. Microbiol. 2014, 5, 409.
- Mayilraj, S.; Kaksonen, A.H.; Cord-Ruwisch, R.; Schumann, P.; Spröer, C.; Tindall, B.J.; Spring, S. Desulfonauticus autotrophicus sp. nov., a novel thermophilic sulfate-reducing bacterium isolated from oil-production water and emended description of the genus Desulfonauticus. Extremophiles 2009, 13, 247–255.
- Orphan, V.; Taylor, L.; Hafenbradl, D.; Delong, E. Culture-dependent and culture-independent characterization of microbial assemblages associated with high-temperature petroleum reservoirs. Appl. Environ. Microbiol. 2000, 66, 700–711.
- Shestakova, N.; Ivoilov, V.; Tourova, T.; Belyaev, S.; Poltaraus, A.; Nazina, T. Application of clone libraries: Syntrophic acetate degradation to methane in a high-temperature petroleum reservoir: Culture-based and 16S rRNA genes characterization. In Applied Microbiology and Molecular Biology in Oil Field Systems; Springer: Cham, Switzerland, 2011; pp. 45–53.
- Mnif, S.; Chamkha, M.; Sayadi, S. Isolation and characterization of Halomonas sp. strain C2SS100, a hydrocarbon-degrading bacterium under hypersaline conditions. J. Appl. Microbiol. 2009, 107, 785–794.
- Cheng, L.; He, Q.; Ding, C.; Dai, L.-R.; Li, Q.; Zhang, H. Novel bacterial groups dominate in a thermophilic methanogenic hexadecane-degrading consortium. FEMS Microbiol. Ecol. 2013, 85, 568–577.
- Mbadinga, S.M.; Li, K.-P.; Zhou, L.; Wang, L.-Y.; Yang, S.-Z.; Liu, J.-F.; Gu, J.-D.; Mu, B.-Z. Analysis of alkane-dependent methanogenic community derived from production water of a high-temperature petroleum reservoir. Appl. Microbiol. Biotechnol. 2012, 96, 531–542.
- Zhou, L.; Li, K.-P.; Mbadinga, S.M.; Yang, S.-Z.; Gu, J.-D.; Mu, B.-Z. Analyses of n-alkanes degrading community dynamics of a high-temperature methanogenic consortium enriched from production water of a petroleum reservoir by a combination of molecular techniques. Ecotoxicology 2012, 21, 1680–1691.
- Pournia, M.; Bahador, N.; Tabatabaei, M.; Azarbayjani, R.; Salekdeh, G.H. Microbial diversity of non-flooded high temperature petroleum reservoir in South of Iran. Biol. J. Microorg. 2020, 8, 15–23.
- Dahle, H.; Garshol, F.; Madsen, M.; Birkeland, N.-K. Microbial community structure analysis of produced water from a high-temperature North Sea oil-field. Antonie Van Leeuwenhoek 2008, 93, 37–49.
- Magot, M.; Ollivier, B.; Patel, B.K.C. Microbiology of petroleum reservoirs. Antonie Van Leeuwenhoek 2000, 77, 103–116.
- Henry, E.A.; Devereux, R.; Maki, J.S.; Gilmour, C.C.; Woese, C.R.; Mandelco, L.; Schauder, R.; Remsen, C.C.; Mitchell, R. Characterization of a new thermophilic sulfate-reducing bacterium. Arch. Microbiol. 1994, 161, 62–69.
- Meslé, M.; Dromart, G.; Oger, P. Microbial methanogenesis in subsurface oil and coal. Res. Microbiol. 2013, 164, 959–972.
- Nazina, T.N.; Shestakova, N.M.; Semenova, E.M.; Korshunova, A.V.; Kostrukova, N.K.; Tourova, T.P.; Min, L.; Feng, Q.; Poltaraus, A.B. Diversity of metabolically active bacteria in water-flooded high-temperature heavy oil reservoir. Front. Microbiol. 2017, 8, 707.
- Silva, T.R.; Verde, L.C.L.; Santos Neto, E.V.; Oliveira, V.M. Diversity analyses of microbial communities in petroleum samples from Brazilian oil fields. Int. Biodeterior. Biodegrad. 2013, 81, 14.
- Liu, J.-F.; Wu, W.-L.; Yao, F.; Wang, B.; Zhang, B.-L.; Mbadinga, S.M.; Gu, J.-D.; Mu, B.-Z. A thermophilic nitrate-reducing bacterium isolated from production water of a high temperature oil reservoir and its inhibition on sulfate-reducing bacteria. Appl. Environ. Biotechnol. 2016, 2, 35–42.
- Tüccar, T.; Ilhan-Sungur, E.; Abbas, B.; Muyzer, G. Coexistence of sulfate reducers with the other oil bacterial groups in Diyarbakır oil fields. Anaerobe 2019, 59, 19–31.
- Nazaries, L.; Murrell, J.C.; Millard, P.; Baggs, L.; Singh, B.K. Methane, microbes and models: Fundamental understanding of the soil methane cycle for future predictions. Environ. Microbiol. 2013, 15, 2395–2417.
- Penger, J.; Conrad, R.; Blaser, M. Stable carbon isotope fractionation by methylotrophic methanogenic archaea. Appl. Environ. Microbiol. 2012, 78, 7596–7602.
- Santos, J.C.D.; Lopes, D.R.G.; Da Silva, J.D.; De Oliveira, M.D.; Dias, R.S.; Lima, H.S.; De Sousa, M.P.; De Paula, S.O.; Silva, C.C.D. Diversity of sulfate-reducing prokaryotes in petroleum production water and oil samples. Int. Biodeterior. Biodegrad. 2020, 151, 104966.
- Summers, Z.M.; Belahbib, H.; Pradel, N.; Bartoli, M.; Mishra, P.; Tamburini, C.; Dolla, A.; Ollivier, B.; Armougom, F. A novel Thermotoga strain TFO isolated from a Californian petroleum reservoir phylogenetically related to Thermotoga petrophila and T. naphthophila, two thermophilic anaerobic isolates from a Japanese reservoir: Taxonomic and genomic considerations. Syst. Appl. Microbiol. 2020, 43, 126132.
- Gieg, L. Microbial communities in oil shales, biodegraded and heavy oil reservoirs, and bitumen deposits. In Microbial Communities Utilizing Hydrocarbons and Lipids: Members, Metagenomics and Ecophysiology; Springer International Publishing: Cham, Switzerland, 2018.
- Qiu, Y.-L.; Hanada, S.; Ohashi, A.; Harada, H.; Kamagata, Y.; Sekiguchi, Y. Syntrophorhabdus aromaticivorans gen. nov., sp. nov., the first cultured anaerobe capable of degrading phenol to acetate in obligate syntrophic associations with a hydrogenotrophic methanogen. Appl. Environ. Microbiol. 2008, 74, 2051–2058.
- Kuever, J. The family Syntrophorhabdaceae. In The Prokaryotes: Deltaproteobacteria and Epsilonproteobacteria; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 301–303.
- Dong, X.; Greening, C.; Rattray, J.E.; Chakraborty, A.; Chuvochina, M.; Mayumi, D.; Dolfing, J.; Li, C.; Brooks, J.M.; Bernard, B.B.; et al. Metabolic potential of uncultured bacteria and archaea associated with petroleum seepage in deep-sea sediments. Nat. Commun. 2019, 10, 1816.
- Aitken, C.M.; Jones, D.M.; Maguire, M.J.; Gray, N.D.; Sherry, A.; Bowler, B.F.J.; Ditchfield, A.K.; Larter, S.R.; Head, I.M. Evidence that crude oil alkane activation proceeds by different mechanisms under sulfate-reducing and methanogenic conditions. Geochim. Cosmochim. Acta 2013, 109, 162–174.
- Davis, J.J.; Wattam, A.R.; Aziz, R.K.; Brettin, T.; Butler, R.; Butler, R.M.; Chlenski, P.; Conrad, N.; Dickerman, A.; Dietrich, E.M.; et al. The PATRIC bioinformatics resource center: Expanding data and analysis capabilities. Nucleic Acids Res. 2020, 48, D606–D612.
- Wang, S.C. Studies of Bacterial Catabolic Enzymes: Implications for the Evolution of Enzymes and Metabolic Pathways. Ph.D. Thesis, The University of Texas, Austin, TX, USA, 2003.
- Davidson, R.; Baas, B.-J.; Akiva, E.; Holliday, G.L.; Polacco, B.J.; LeVieux, J.A.; Pullara, C.R.; Zhang, Y.J.; Whitman, C.P.; Babbitt, P.C. A global view of structure–function relationships in the tautomerase superfamily. J. Biol. Chem. 2018, 293, 2342–2357.
- Michał, B.; Gagat, P.; Jabłoński, S.; Chilimoniuk, J.; Gaworski, M.; Mackiewicz, P.; Marcin, Ł.; Burdukiewicz, M.; Łukaszewicz, M. PhyMet2: A database and toolkit for phylogenetic and metabolic analyses of methanogens. Environ. Microbiol. Rep. 2018, 10, 378–382.
- Borrel, G.; Adam, P.S.; McKay, L.J.; Chen, L.-X.; Sierra-García, I.N.; Sieber, C.M.K.; Letourneur, Q.; Ghozlane, A.; Andersen, G.L.; Li, W.-J.; et al. Wide diversity of methane and short-chain alkane metabolisms in uncultured archaea. Nat. Microbiol. 2019, 4, 603–613.
- Laso-Pérez, R.; Hahn, C.; Vliet, D.M.V.; Tegetmeyer, H.E.; Schubotz, F.; Smit, N.T.; Pape, T.; Sahling, H.; Bohrmann, G.; Boetius, A.; et al. Anaerobic degradation of non-methane alkanes by candidatus methanoliparia in hydrocarbon seeps of the gulf of Mexico. mBio 2019, 10, e01814–e01819.
- Lahme, S.; Callbeck, C.M.; Eland, L.E.; Wipat, A.; Enning, D.; Head, I.M.; Hubert, C.R.J. Comparison of sulfide-oxidizing Sulfurimonas strains reveals a new mode of thiosulfate formation in subsurface environments. Environ. Microbiol. 2020, 22, 1784–1800.
- Dahle, H.; Roalkvam, I.; Thorseth, I.H.; Pedersen, R.B.; Steen, I.H. The versatile in situ gene expression of an E psilonproteobacteria-dominated biofilm from a hydrothermal chimney. Environ. Microbiol. Rep. 2013, 5, 282–290.
- Hensen, D.; Sperling, D.; Trüper, H.G.; Brune, D.C.; Dahl, C. Thiosulphate oxidation in the phototrophic sulphur bacterium Allochromatium vinosum. Mol. Microbiol. 2006, 62, 794–810.
- Zumft, W.; Kroneck, P. Respiratory transformation of nitrous oxide (N2O) to dinitrogen by bacteria and archaea. In Advances in Microbial Physiology; Poole, R.K., Ed.; Academic Press: Cambridge, MA, USA, 2007; Volume 52.

