 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sumit Mukherjee | + 1768 word(s) | 1768 | 2021-08-31 04:56:18 | | | |
| 2 | Enzi Gong | Meta information modification | 1768 | 2021-09-02 03:02:54 | | |
Video Upload Options
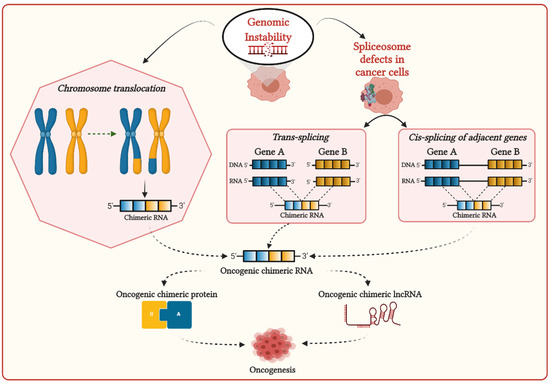
Due to the consequences of genome instability in cancer cells, sometimes two mRNAs can be fused to generate chimeric RNAs. Several recent studies demonstrated that chimeric RNAs are significantly associated with oncogenesis and can also promote drug resistance. The generation of chimeric RNAs could allow cancer cells to switch their functionality. Therefore, chimeric RNAs are an important driver for generating the phenotypic plasticity of cancer cells and increasing their fitness in the tissue environment. Chimeric RNAs could be translated and generate new fusion or chimeric proteins that could alter the normal pathways and lead to cancer development. Chimeric RNAs could also generate long non-coding RNA (lncRNA), which could regulate cancer cell proliferation.
1. Overview
Fusion of exons or introns from two different genes can lead to the formation of chimeric RNAs. Several recent studies have reported that chimeric RNAs promote tumorigenesis and cancer drug resistance. Therefore, chimeric RNAs are crucial for generating phenotypic diversity between cancer cells that drives the adaptive evolution of cancer.
Gene fusions can give rise to somatic alterations in cancers. Fusion genes have the potential to create chimeric RNAs, which can generate the phenotypic diversity of cancer cells, and could be associated with novel molecular functions related to cancer cell survival and proliferation. The expression of chimeric RNAs in cancer cells might impact diverse cancer-related functions, including loss of apoptosis and cancer cell plasticity, and promote oncogenesis. Due to their recurrence in cancers and functional association with oncogenic processes, chimeric RNAs are considered biomarkers for cancer diagnosis. Several recent studies demonstrated that chimeric RNAs could lead to the generation of new functionality for the resistance of cancer cells against drug therapy. Therefore, targeting chimeric RNAs in drug resistance cancer could be useful for developing precision medicine. So, understanding the functional impact of chimeric RNAs in cancer cells from an evolutionary perspective will be helpful to elucidate cancer evolution, which could provide a new insight to design more effective therapies for cancer patients in a personalized manner.
2. Cancer Evolution
3. Mechanisms of Formation of Chimeric RNAs in Cancer Cells and Their Functional Associations with Cancer Development
4. Conclusions
References
- Black, J.R.M.; McGranahan, N. Genetic and non-genetic clonal diversity in cancer evolution. Nat. Rev. Cancer 2021, 21, 379–392.
- Yates, L.R.; Campbell, P.J. Evolution of the cancer genome. Nat. Rev. Genet. 2012, 13, 795–806.
- Horne, S.D.; Pollick, S.A.; Heng, H.H.Q. Evolutionary mechanism unifies the hallmarks of cancer. Int. J. Cancer 2015, 136, 2012–2021.
- Heng, J.; Heng, H.H. Genome chaos: Creating new genomic information essential for cancer macroevolution. Semin. Cancer Biol. 2020.
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313.
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345.
- Birkbak, N.J.; McGranahan, N. Cancer Genome Evolutionary Trajectories in Metastasis. Cancer Cell 2020, 37, 8–19.
- Heng, H.H. Genome Chaos: Rethinking Genetics, Evolution, and Molecular Medicine; Academic Press Elsevier: Cambridge, MA, USA, 2019.
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337.
- Marjanovic, N.D.; Weinberg, R.A.; Chaffer, C.L. Cell plasticity and heterogeneity in cancer. Clin. Chem. 2013, 59, 168–179.
- Barber, L.J.; Davies, M.N.; Gerlinger, M. Dissecting cancer evolution at the macro-heterogeneity and micro-heterogeneity scale. Curr. Opin. Genet. Dev. 2015, 30, 1–6.
- Xu, Y.; Li, H.; Yang, F.; Yang, D.; Zhou, B.-B.S. Cell plasticity and genomic instability in cancer evolution. Genome Instab. Dis. 2020, 1, 301–309.
- Shapiro, J.A. How chaotic is genome chaos? Cancers 2021, 13, 1358.
- Heng, H.H.; Bremer, S.W.; Stevens, J.B.; Horne, S.D.; Liu, G.; Abdallah, B.Y.; Ye, K.J.; Ye, C.J. Chromosomal instability (CIN): What it is and why it is crucial to cancer evolution. Cancer Metastasis Rev. 2013, 32, 325–340.
- Lee, J.K.; La Choi, Y.; Kwon, M.; Park, P.J. Mechanisms and Consequences of Cancer Genome Instability: Lessons from Genome Sequencing Studies. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 283–312.
- Wei, D.; Yao, Y. Genomic Instability and Cancer. J. Carcinog. Mutagen. 2014, 5, 1000165.
- Li, Z.; Qin, F.; Li, H. Chimeric RNAs and their implications in cancer. Curr. Opin. Genet. Dev. 2018, 48, 36–43.
- Wu, H.; Li, X.; Li, H. Gene fusions and chimeric RNAs, and their implications in cancer. Genes Dis. 2019, 6, 385–390.
- Gao, Q.; Liang, W.W.; Foltz, S.M.; Mutharasu, G.; Jayasinghe, R.G.; Cao, S.; Liao, W.W.; Reynolds, S.M.; Wyczalkowski, M.A.; Yao, L.; et al. Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep. 2018, 23, 227–238.e3.
- Turner, N.C.; Reis-Filho, J.S. Genetic heterogeneity and cancer drug resistance. Lancet Oncol. 2012, 13, e178–e185.
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309.
- Qin, S.; Jiang, J.; Lu, Y.; Nice, E.C.; Huang, C.; Zhang, J.; He, W. Emerging role of tumor cell plasticity in modifying therapeutic response. Signal. Transduct. Target. Ther. 2020, 5, 1–36.
- Frenkel-Morgenstern, M.; Gorohovski, A.; Tagore, S.; Sekar, V.; Vazquez, M.; Valencia, A. ChiPPI: A novel method for mapping chimeric protein-protein interactions uncovers selection principles of protein fusion events in cancer. Nucleic Acids Res. 2017, 45, 7094–7105.
- Latysheva, N.S.; Babu, M.M. Molecular Signatures of Fusion Proteins in Cancer. ACS Pharmacol. Transl. Sci. 2019, 2, 122–133.
- Qin, F.; Zhang, Y.; Liu, J.; Li, H. SLC45A3-ELK4 functions as a long non-coding chimeric RNA. Cancer Lett. 2017, 404, 53–61.
- Mukherjee, S.; Detroja, R.; Balamurali, D.; Matveishina, E.; Medvedeva, Y.A.; Valencia, A.; Gorohovski, A.; Frenkel-Morgenstern, M. Computational analysis of sense-antisense chimeric transcripts reveals their potential regulatory features and the landscape of expression in human cells. NAR Genom. Bioinform. 2021, 3.
- Mitelman, F.; Johansson, B.; Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 2007, 7, 233–245.
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer 2015, 15, 371–381.
- Brien, G.L.; Stegmaier, K.; Armstrong, S.A. Targeting chromatin complexes in fusion protein-driven malignancies. Nat. Rev. Cancer 2019, 19, 255–269.
- Rowley, J.D. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973, 243, 290–293.
- Shtivelman, E.; Lifshitz, B.; Gale, R.P.; Canaani, E. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature 1985, 315, 550–554.
- Collins, S.J. Retinoic acid receptors, hematopoiesis and leukemogenesis. Curr. Opin. Hematol. 2008, 15, 346–351.
- Zheng, J. Oncogenic chromosomal translocations and human cancer (Review). Oncol. Rep. 2013, 30, 2011–2019.
- Saeed, S.; Logie, C.; Stunnenberg, H.G.; Martens, J.H.A. Genome-wide functions of PML-RARα in acute promyelocytic leukaemia. Br. J. Cancer 2011, 104, 554–558.
- Clark, J.P.; Cooper, C.S. ETS gene fusions in prostate cancer. Nat. Rev. Urol. 2009, 6, 429–439.
- White, N.M.; Feng, F.Y.; Maher, C.A. Recurrent Rearrangements in Prostate Cancer: Causes and Therapeutic Potential. Curr. Drug Targets 2013, 14, 450–459.
- Tomlins, S.A.; Laxman, B.; Varambally, S.; Cao, X.; Yu, J.; Helgeson, B.E.; Cao, Q.; Prensner, J.R.; Rubin, M.A.; Shah, R.B.; et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia 2008, 10, 177-IN9.
- Hessels, D.; Schalken, J.A. Recurrent gene fusions in prostate cancer: Their clinical implications and uses. Curr. Urol. Rep. 2013, 14, 214–222.
- Haluska, F.G.; Tsujimoto, Y.; Croce, C.M. The t(8;14) chromosome translocation of the Burkitt lymphoma cell line Daudi occurred during immunoglobulin gene rearrangement and involved the heavy chain diversity region. Proc. Natl. Acad. Sci. USA 1987, 84, 6835–6839.
- Boerma, E.G.; Siebert, R.; Kluin, P.M.; Baudis, M. Translocations involving 8q24 in Burkitt lymphoma and other malignant lymphomas: A historical review of cytogenetics in the light of todays knowledge. Leukemia 2009, 23, 225–234.
- Djabali, M.; Selleri, L.; Parry, P.; Bower, M.; Young, B.D.; Evans, G.A. A trithorax–like gene is interrupted by chromosome 11q23 translocations in acute leukaemias. Nat. Genet. 1992, 2, 113–118.
- Ziemin-Van Der Poel, S.; Mccabe, N.R.; Gill, H.J.; Espinosa, R.; Patel, Y.; Harden, A.; Rubinelli, P.; Smith, S.D.; Lebeau, M.M.; Rowley, I.D.; et al. Identification of a gene, MLL, that spans the breakpoint in 11q23 translocations associated with human leukemias. Proc. Natl. Acad. Sci. USA 1991, 88, 10735–10739.
- Krivtsov, A.V.; Armstrong, S.A. MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 2007, 7, 823–833.
- Andersson, A.K.; Ma, J.; Wang, J.; Chen, X.; Gedman, A.L.; Dang, J.; Nakitandwe, J.; Holmfeldt, L.; Parker, M.; Easton, J.; et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat. Genet. 2015, 47, 330–337.
- Xu, J.; Li, L.; Xiong, J.; DenDekker, A.; Ye, A.; Karatas, H.; Liu, L.; Wang, H.; Qin, Z.S.; Wang, S.; et al. MLL1 and MLL1 fusion proteins have distinct functions in regulating leukemic transcription program. Cell Discov. 2016, 2, 16008.
- Parameswaran, S.; Vizeacoumar, F.S.; Kalyanasundaram Bhanumathy, K.; Qin, F.; Islam, M.F.; Toosi, B.M.; Cunningham, C.E.; Mousseau, D.D.; Uppalapati, M.C.; Stirling, P.C.; et al. Molecular characterization of an MLL1 fusion and its role in chromosomal instability. Mol. Oncol. 2019, 13, 422–440.
- Rinaldi, C.; Pizzul, P.; Longhese, M.P.; Bonetti, D. Sensing R-Loop-Associated DNA Damage to Safeguard Genome Stability. Front. Cell Dev. Biol. 2021, 8, 1657.
- Nguyen, H.D.; Leong, W.Y.; Li, W.; Reddy, P.N.G.; Sullivan, J.D.; Walter, M.J.; Zou, L.; Graubert, T.A. Spliceosome mutations induce R loop-associated sensitivity to ATR inhibition in myelodysplastic syndromes. Cancer Res. 2018, 78, 5363–5374.
- Chen, L.; Chen, J.Y.; Huang, Y.J.; Gu, Y.; Qiu, J.; Qian, H.; Shao, C.; Zhang, X.; Hu, J.; Li, H.; et al. The Augmented R-Loop Is a Unifying Mechanism for Myelodysplastic Syndromes Induced by High-Risk Splicing Factor Mutations. Mol. Cell 2018, 69, 412–425.e6.
- Boros-Oláh, B.; Dobos, N.; Hornyák, L.; Szabó, Z.; Karányi, Z.; Halmos, G.; Roszik, J.; Székvölgyi, L. Drugging the R-loop interactome: RNA-DNA hybrid binding proteins as targets for cancer therapy. DNA Repair 2019, 84, 102642.
- Tam, A.S.; Stirling, P.C. Splicing, genome stability and disease: Splice like your genome depends on it! Curr. Genet. 2019, 65, 905–912.
- Jia, Y.; Xie, Z.; Li, H. Intergenically Spliced Chimeric RNAs in Cancer. Trends Cancer 2016, 2, 475–484.
- Grosso, A.R.; Leite, A.P.; Carvalho, S.; Matos, M.R.; Martins, F.B.; Vítor, A.C.; Desterro, J.M.P.; Carmo-Fonseca, M.; de Almeida, S.F. Pervasive transcription read-through promotes aberrant expression of oncogenes and RNA chimeras in renal carcinoma. eLife 2015, 4, e09214.
- Nacu, S.; Yuan, W.; Kan, Z.; Bhatt, D.; Rivers, C.S.; Stinson, J.; Peters, B.A.; Modrusan, Z.; Jung, K.; Seshagiri, S.; et al. Deep RNA sequencing analysis of readthrough gene fusions in human prostate adenocarcinoma and reference samples. BMC Med. Genom. 2011, 4, 11.
- Varley, K.E.; Gertz, J.; Roberts, B.S.; Davis, N.S.; Bowling, K.M.; Kirby, M.K.; Nesmith, A.S.; Oliver, P.G.; Grizzle, W.E.; Forero, A.; et al. Recurrent read-through fusion transcripts in breast cancer. Breast Cancer Res. Treat. 2014, 146, 287–297.
- Maher, C.A.; Kumar-Sinha, C.; Cao, X.; Kalyana-Sundaram, S.; Han, B.; Jing, X.; Sam, L.; Barrette, T.; Palanisamy, N.; Chinnaiyan, A.M. Transcriptome sequencing to detect gene fusions in cancer. Nature 2009, 458, 97–101.
- Rickman, D.S.; Pflueger, D.; Moss, B.; VanDoren, V.E.; Chen, C.X.; De la Taille, A.; Kuefer, R.; Tewari, A.K.; Setlur, S.R.; Demichelis, F.; et al. SLC45A3-ELK4 is a novel and frequent erythroblast transformation-specific fusion transcript in prostate cancer. Cancer Res. 2009, 69, 2734–2738.
- Li, H.; Wang, J.; Mor, G.; Sklar, J. A neoplastic gene fusion mimics trans-splicing of RNAs in normal human cells. Science 2008, 321, 1357–1361.
- Gingeras, T.R. Implications of chimaeric non-co-linear transcripts. Nature 2009, 461, 206–211.
- Yuan, H.; Qin, F.; Movassagh, M.; Park, H.; Golden, W.; Xie, Z.; Zhang, P.; Sklar, J.; Li, H. A chimeric RNA characteristic of rhabdomyosarcoma in normal myogenesis process. Cancer Discov. 2013, 3, 1394–1403.


