Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | MARCO LANA-PEIXOTO | + 2977 word(s) | 2977 | 2021-07-07 05:53:55 | | | |
| 2 | Vivi Li | Meta information modification | 2977 | 2021-09-01 04:22:01 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Lana-Peixoto, M. Neuromyelitis Optica Spectrum Disorder. Encyclopedia. Available online: https://encyclopedia.pub/entry/13749 (accessed on 06 June 2026).
Lana-Peixoto M. Neuromyelitis Optica Spectrum Disorder. Encyclopedia. Available at: https://encyclopedia.pub/entry/13749. Accessed June 06, 2026.
Lana-Peixoto, Marco. "Neuromyelitis Optica Spectrum Disorder" Encyclopedia, https://encyclopedia.pub/entry/13749 (accessed June 06, 2026).
Lana-Peixoto, M. (2021, August 31). Neuromyelitis Optica Spectrum Disorder. In Encyclopedia. https://encyclopedia.pub/entry/13749
Lana-Peixoto, Marco. "Neuromyelitis Optica Spectrum Disorder." Encyclopedia. Web. 31 August, 2021.
Copy Citation
The term neuromyelitis optica was introduced by Eugène Devic and Fernand Gault in 1894, who first recognized the association of amaurosis and myelitis as a new clinical entity.
neuromyelitis optica spectrum disorders
anti-MOG syndrome
aquaporin 4-IgG
myelin oligodendrocyte glycoprotein
multiple sclerosis
1. Neuromyelitis Optica Spectrum Disorders
The term neuromyelitis optica was introduced by Eugène Devic and Fernand Gault in 1894, who first recognized the association of amaurosis and myelitis as a new clinical entity. Devic [1] reported the case of a 45-year-old French woman who was seen at the Hôtel-Dieu hospital of Lyon because of an intractable headache and depression in addition to general asthenia. One month later, she developed urinary retention, complete paraplegia and blindness, and died few weeks later. Autopsy disclosed severe demyelinating and necrotic lesions extending 4–5 cm length in the lower thoracic and lumbar spinal cord. There was demyelination of the optic nerves, but a gross examination of the brain was unrevealing. In this paper, Devic emphasized the similarity of the pathological process involving the spinal cord and the optic nerves, named the syndrome “neuro-myélite optique”, or “neuroptico-myélite”, and discussed its relationship with MS. Fernand Gault, a disciple of Devic’s, reviewed in detail 17 cases of this condition in his doctoral thesis named “De la neuromyélite optique aiguë” [2]. The eponym “Devic’s disease” was suggested by Acchiote [3]. However, the association of myelitis and blindness had already been reported by other authors in the early and mid 19th century. The case of Marquis de Causan—known as the first description of this association by the French anatomist and pathologist Antoine Portal in 1803–1804—was characterized by relapsing myelitis followed by amaurosis and signs of brainstem involvement [4]. Other previously reported cases included those by Giovanni Battista Pescetto in 1844 [5], Christopher Mercer Durrant in 1850 [6], Jacob Augustus Lockhart Clarke in 1862 [7], Thomas Clifford Albutt in 1870 [8], and Wilhelm Heinrich Erb in 1879–1880 [9]. Also, in the American continent, the association of optic neuritis and myelitis was identified by Seguin (1880) [10] prior to Devic and Gault’s pioneering publication. None of these previous authors however, had used the term “neuromyelitis optica”, or considered their cases as expression of a new nosological entity. It was only in 1943 that the disease was first identified in Latin America, when Aluizio Marques, in Rio de Janeiro, described two female patients who developed bilateral blindness and acute transverse myelitis [11].
2. Pathophysiology
The discovery of NMO-IgG and AQP4 as its targeted antigen unequivocally confirmed neuromyelitis optica as a disease distinct from MS and allowed its early laboratorial recognition [12][13]. The serum identification of AQP4-IgG expanded the clinical spectrum of the disease to include its limited forms (single or recurrent longitudinally extensive transverse myelitis [LETM], defined by MRI as a lesion extending for three or more vertebral segments, or recurrent isolated optic neuritis) [14], along with a wide variety of brainstem, diencephalic, and cerebral manifestations [15][16].
Aquaporin-4 monomers assemble to form tetramers which further aggregate in cell plasma membranes to form supramolecular arrays called orthogonal arrays of particles (OAP). There are two major forms of AQP-4: the full-length 323 amino acid M1 isoform, and the shorter 301 amino acid M23 isoform. Only the M23 isoform forms large OAPs [17]. It has been shown that M1 isoform does not form OAPs on its own, but can co-assemble with M23 in heterotetramers that limit OAP size [18][19].
Aquaporin-4 is widely expressed throughout the CNS. It is also highly expressed in the optic nerves and spinal cord, explaining their preferential involvement in the disease. Other CNS sites expressing AQP-4 include the supraoptic nucleus of the hypothalamus, the periventricular structures such as area postrema and the vascular organ of lamina terminalis, which lack BBB and contain osmo-sensitive neurons that regulate fluid homeostasis and release arginine-vasopressin, which facilitates this process. Aquaporin-4 is also expressed in non-neural tissues including skeletal muscle cells, lung airway cells, gastric parietal cells, renal collecting duct cells, inner ear, retinal Muller cells, lacrimal gland and salivary duct cells, and olfactory epithelial cells [20]. Human and experimental studies have shown that AQP4-IgG belongs mainly to IgG1 class, a potent activator of complement. The antibody enters the CNS, binds the antigen at astrocyte processes, induces complement-mediated inflammation, granulocyte infiltration, and astrocyte death [19][21]. Complement-mediated inflammation with secondary neutrophils and eosinophil infiltration plays a key role in the pathophysiology of NMOSD attacks [22].
Aquaporin-4 antibodies are more abundant in the peripheral blood than in the cerebrospinal fluid (CSF) [23]. In the periphery, they are produced by a number of B cell subpopulations which are vulnerable to interleukin-6 (IL-6), which enhances their survival and AQP4-IgG secretion [24]. Although AQP4-IgG can gain direct access to AQP4 on astrocytes located at circumventricular organs where the endothelia lack tight junction, the mechanisms of its penetration into other CNS sites that are protected byBBB are still unclear. Recent findings have shown that AQP4-IgG is not sufficient or even necessary to cause BBB disruption [25]. Sera from NMO patients contain non-reactive AQP4 antibodies, identified as recombinant antibodies (rAb) ON-12-2-46 and ON-07-5-31 which target glucose-regulated protein 78 (GRP78) on the cell surface of brain microvascular endothelial cells (BMEC). GRP78 is a stress protein of the heat shock 70 family expressed in all CNS cells [26]. However, only rAb ON 12-2-46 induces nuclear translocation of nuclear factor kB (NF-kB) p65, which is a marker of cell activation [25]. BMECs activation causes increased secretion of vascular endothelium growth factor (VEGF) and metaloproteinases (MMP)-2/9 which result in a down regulation of claudin 5 and disruption of the BBB. Leakage of the BBB allows entrance of AQP4-IgG to the CNS and its binding to AQP4 in the astrocytic endfeet. Evidences showing the causal role of AQP4-IgG in NMOSD include its nearly absolute specificity for the disease; its correlation with disease activity, higher number of relapses and more severe course as compared with seronegative patients; some distinct demographic and clinical features; increased concentration of AQP4-positive plasmablasts in NMOSD patients, mainly during disease relapses; and decreased serum AQP4-IgG concentration following successful treatments and during disease remission [27][28][29]. Histopathological features such as a marked loss of astrocytes and accumulation of IgG and IgM around blood vessels, the site of AQP4 expression; spare of myelin and axons in some lesions suggesting that astrocytes (which have a higher expression of AQP4) are the initial cell target in the disease, whereas in more recent lesions there may be preservation of glial fibrillary acidic protein (GFAP), a marker of astrocyte damage, suggesting that AQP4 is the primary target of the immune attack. The initial loss of AQP4 with astrocyte preservation might reflect the internalization of AQP4 of either M1-AQP4 or both isoforms before a complement becomes locally available to mediate a lytic inflammatory process. This would account for the rapid reversal of some MRI abnormalities in the area postrema and some parts of the cerebrum. The disease individual clinical phenotype and severity may be related to the ratio of AQP4-M1 to M23 in the optic nerves, brain, and spinal cord. While CNS regions with a higher proportion of M1 would rapidly internalize, avoiding a cascade of tissue damage, areas richer in AQP4-M23 isoform would be more liable to necrotic lesions and cavitation [26][27][28].
Additionally, following a decreased serum concentration of AQP4-IgG, the AQP4-M1 isoform is rapidly replaced in the astrocytic membrane. Experimental studies have showed that a passive transfer of AQP4-IgG from NMOSD patients to animals with disrupted BBB by previous experimental autoimmune encephalomyelitis (EAE), or through pretreatment with Freund’s adjunct, develop CNS typical NMO histopathological lesions [19][30][31][32][33].
3. Epidemiology
Some caveats are needed before looking at published data on the epidemiology of NMOSD. First, studies on the frequency and distribution of the disease across the world are still scanty and most of them are based on small cohorts. Additionally, they employed non-standardized methodology and inconsistent inclusion of seronegative patients, and differ regarding used diagnostic criteria and assays for AQP4 antibodies detection. In spite of these limitations, a number of issues related to disease prevalence, ethnicity and geography have been clarified. Studies in different populations and geographic regions show that NMOSD is a rare disorder with worldwide distribution [34]. However, an exception to these studies is the seroprevalence study from Olmsted County, in the United States, and Martinique [35] which showed a 2.5-fold higher prevalence rate of the disease in Martinique (10 per 100,000) than in Olmsted County (3.9 per 100,000), all other studies [34] indicated a fairly uniform prevalence rate below 5 per 100,000 people in different regions and populations. Likewise, incidence rates were also homogeneous in different countries, ranging from 0.2 per 1 million per year, in Mexico [36] to 4.0 in Denmark [37]. Although most studies point to incidence rate below 1 per 1 million, a peak incidence rate of 7.3 per 1 million was again found in Martinique [35].
Although NMOSD has been regarded to have a predilection for non-Caucasians, the similarity of prevalence and incidence rates in different geographic regions and distinct ethnicities may suggest that, as in opposition to MS, latitude and genetic factors may not play a key role in NMOSD pathogenesis [38]. This contradicts the ethnicity-specific higher prevalence for blacks in Olmsted County, which was similar to that found in the black population of Martinique [35]. The Australian-New Zealand study also observed higher prevalence rate in people with Asian ancestry than in Caucasians [39].
Ethnicity, however, influences age at onset and phenotype of AQP4 seropositive NMOSD. A recent study comparing the clinical manifestations and outcome of 603 NMOSD patients of three different races (Asians, Caucasians and Afro-Americans/Afro-Europeans) showed that non-white patients are younger at disease onset, and more frequently have brain attacks at onset or during the disease course, as well as more frequent abnormalities on brain MRI. Afro-American and Afro-European patients have more severe attacks at onset than Asians and Caucasians, but the outcome at the last follow-up was similar in the different racial groups [40]. This observed that the similarity of the outcome at the last follow-up for all racial groups is in opposition to previous reports of a more severe outcome of NMOSD in Afro-Caribbean than in Caucasian patients [41][42][43].
Usually, the initial clinical manifestations of NMOSD occur at an age of around 35-45 years (median age at onset is 39), but children and the elderly account for 18% of cases. Women comprise 70% to 90% of all cases, but there is no gender predilection in children [44]. The estimated proportion of familial cases (3%) is greater than expected based on the disease prevalence [45]. In some populations, human leukocyte antigen (HLA) has been reported to be associated with susceptibility to NMOSD, such as HLA-DPB1*0501 allele in Japanese and Chinese populations [46][47] and HLA-DPB1*03 in Caucasian, Afro-Caribbean, and Indian patients [48][49][50][51]. These genetic factors may account for the phenotypic variability among racial groups.
4. Magnetic Resonance Imaging
Magnetic resonance imaging of the brain and spinal cord is an essential tool for the diagnosis and management of demyelinating diseases of the CNS. The correct differentiation of NMOSD and anti-MOG syndromes from MS is important to provide patients with the most appropriate treatment.
Longitudinally extensive transverse myelitis, is the most specific imaging feature of NMOSD (Figure 1a). The length of the lesion has been considered the most distinguishing feature from MS, although long lesions may occur in MS and short lesions in NMOSD. Frequently, LETM lesions exhibit non-homogeneous contrast-enhancing that may persists for months following acute attacks. An extensive centrally-located hypointense signal in T1-sequence denotes cavitation secondary to tissue necrosis (Figure 1b). Cervical lesions may extend rostrally to the medulla oblongata (Figure 1c). Longitudinally extensive cord atrophy results from severe or recurring myelitis (Figure 1d). Short lesions, characterized by extension < three vertebral segments have been reported, predominantly at disease onset in 14% of the patients [52].
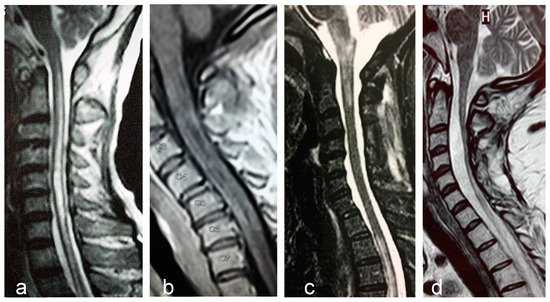
Figure 1. Examples of longitudinally extensive spinal cord lesions detected by MRI in AQP4- seropositive NMOSD patients. (a). T2-weighted central longitudinally extensive cervical lesion. (b). T1-weighted lesion with gadolinium showing multiple hypointensities (cavitations) throughout the cervical cord. (c). T2-weighted cervical lesion extending to brainstem. Another lesion is seen in the upper thoracic levels. (d). Longitudinally extensive spinal cord atrophy of the cervical cord.
Optic nerve abnormalities differ between, NMOSD and MS. Thickened, contrast-enhancing and long (≥ one-half the length of the optic nerve) lesions, as well as preference for involvement of the posterior segment of the nerve or chiasm are all in favor of NMOSD (Figure 2).
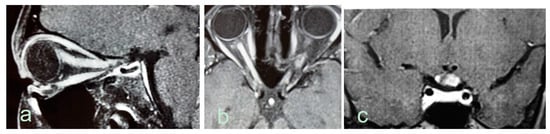
Figure 2. Optic nerve abnormalities on MRI in AQP4-seropositive NMOSD patients. (a). Sagittal T1-weighted MRI shows edematous gadolinium-enhancing optic nerve lesion extending from the eye to the intracranial segment. (b). Axial T1-weighted extensive gadolinium-enhancing lesion in both optic nerves. (c). Coronal T1-weighted MRI shows edematous gadolinium enhancing lesion in the optic chiasm.
Most NMOSD patients have abnormalities on brain MRI [53]. More commonly, brain MRI lesions are unspecific, but they fulfill Barkoff’s criteria for MS in up to 42% of patients [53][54]. In a minority of cases, NMOSD typical brain lesions can be identified mainly in AQP4 enriched regions, such as around the lateral, third and fourth ventricles [53]. Brain lesions that favor NMOSD more than MS include peri-ependymal lesions surrounding the ventricles and aqueduct, hemispheric tumefactive lesions, extensive lesions involving corticospinal tracts, and “cloud-like” enhancing lesions [55]. One recent study [56] showed that criteria comprising (1) at least one lesion adjacent to the body of the lateral ventricle and in the inferior temporal lobe; or (2) the presence of an S-shaped U-fiber lesion; or (3) a Dawson’s finger type lesion were fulfilled by 90.9% RRMS, 12.9% AQP4-IgG NMOSD, and 4.8% MOG-IgG NMOSD patients.
Adults and children with MOG antibody disease frequently had fluffy brainstem lesions, often located in pons and/or adjacent to fourth ventricle. Children across all conditions showed more frequent bilateral, large, brainstem and deep grey matter lesions. MOG antibody disease spontaneously separated from multiple sclerosis, but overlapped with AQP4 antibody disease. Multiple sclerosis was discriminated from MOG antibody disease and from AQP4 antibody disease with high predictive values, while MOG antibody disease could not be accurately discriminated from AQP4 antibody disease. The best classifiers between MOG antibody disease and multiple sclerosis were similar in adults and children, and included ovoid lesions adjacent to the body of lateral ventricles, Dawson’s fingers T1 hypointense lesions (multiple sclerosis), fluffy lesions and three lesions or less (MOG antibody). In the validation cohort patients with antibody-mediated conditions were differentiated from multiple sclerosis with high accuracy [56].
5. Treatment
In spite of their clinical similarities, NMOSD and MS have different treatment. It has been shown that most MS disease modifying drugs, including beta-interferons, glatiramer acetate, natalizumab, alemtuzumab, fingolimod and dimethyl-fumarate are not only inefficacious in NMOSD, but may cause disease exacerbation [57].
In NMOSD the outcome of attacks is usually poor. Recent analysis of 871 attacks revealed that complete remission occurred in only 21% of them and 6% of them had no improvement [58]. The sequence of treatments is of fundamental importance to improve the outcome. Medical therapy, therefore, aims both to enfeeble an ongoing inflammatory attack, and avoid future relapses.
5.1. Therapy of Acute Relapses
Relapses are usually treated with intravenous pulses of methylprednisolone (one gram/day for five days). In severe NMOSD attacks, or when corticosteroids fail to stabilize progression of symptoms plasma exchange (PLEX) must be added [59]. Apheresis eliminates the pathogenic antibodies, from circulation and has higher therapeutic efficacy than IV corticosteroids. Its early use as first-line therapy following attack is a predictor of better remission.
Post-infusion oral prednisone is usually recommended, mainly when an immunosuppressive agent with delayed onset of action is prescribed as prophylaxis of new events [60]. Azathioprine, mycophenolate mofetil, and rituximab are the most commonly used immunosuppressive treatments for prevention of new attacks of the disease.
5.2. Therapy for Relapses Prevention
Azathioprine is probably the most commonly used drug in the preventive treatment of attacks in NMOSD. Initially, it should be combined with prednisone for three to six months until its maximal therapeutic effect can be reached. The lymphocyte count should decrease to 600–1000/cubic millimeter and the mean erythrocyte volume should increase five points from baseline. Thiopurine methyltransferase enzyme activity testing, when available is recommended before the administration of the drug to avoid higher risk of adverse effects. Monitoring of blood cell count and liver function tests on a regular basis is mandatory.
Mycophenolate mofetil is recommended as an alternative treatment in patients who develop intolerance or poor response to azathioprine.
Rituximab is a chimeric monoclonal anti-CD20 antibody that produces rapid depletion of circulating CD20 B cells. A number of studies have showed its efficacy and tolerance in the treatment of NMOSD, but some aspects of treatment strategy and long-term safety still remain to be clarified [61].
Monoclonal antibodies will probably play a most important role in treatment of NMOSD in the coming years. Eculizumab and tocilizumab have already shown their efficacy in small groups of patients [62].
Eculizumab is a humanized monoclonal antibody that inhibits the complement protein C5 and blocks terminal complement activation [63]. The complement cascade is a fundamental part in the inflammation process in NMOSD lesions. In spite of eculizumab efficacy in preventing relapses the increased risk of patients developing meningococcal meningitis raises important safety concerns [64].
Tocilizumab is a monoclonal antibody that targets Interleukin-6 (IL-6) receptor and decrease survival of the antibody-producing plasmablasts. Inebelizumab is a humanized anti-CD19 monoclonal antibody that targets B cell lineage. Although there is still no open-label study supporting its use, it is probably more efficacious than rituximab, which targets the more mature CD20. Inebelizumab removes plasmablasts that express CD19, decreasing the production of AQP4-IgG [62].
Satralizumab is an anti-IL-6 receptor monoclonal antibody. A recent communication on results of a phase III study showed that Satralizumab is a promising therapeutic agent by reducing the risk of relapses by 62% in NMOSD patients [65].
Tolerization is a recent therapeutic approach that uses innovative techniques to restore immune tolerance to host antigens and suppress autoimmune diseases [66]. Tolerization techniques include inverse DNA vaccination, T-cell vaccination, peptide-coupling strategies, tolerogenic dendritic cell vaccination, as well as T-cell receptor engineering-, and chimeric antigen receptor-based therapeutics. As AQP-4 is a specific target to NMO-IgG, there is reason for optimism that this new approach might offer marked beneficial to NMOSD patients, avoiding the wide variety of adverse effects of chronic immunosuppressive agents.
References
- Devic, E. Myelite subaigue compliquee de nevrite optique. Bull. Med. 1894, 8, 1033.
- Gault, F. De la neuromyélite optique aiguë. Ph.D. Thesis, Alexandre Rey, imprimeur de la faculté de médecine, Faculté de Medicine et de Pharmacie de Lyon, Lyon, France, 1894.
- Acchiote, P. Sur un cas de neuromyélite subaiguë ou maladie de devic. Rev. Neurol. 1907, 20, 775–777.
- Jarius, S.; Wildemann, B. The case of the marquis de causan (1804): An early account of visual loss associated with spinal cord inflammation. J. Neurol. 2012, 259, 1354–1357.
- Jarius, S.; Wildemann, B. ‘Noteomielite’ accompanied by acute amaurosis (1844). An early case of neuromyelitis optica. J. Neurol. Sci. 2012, 313, 182–184.
- Jarius, S.; Wildemann, B. An early british case of neuromyelitis optica (1850). BMJ Clin. Res. Ed. 2012, 345, e6430.
- Jarius, S.; Wildemann, B. An early case of neuromyelitis optica: On a forgotten report by jacob lockhart clarke, frs. Mult. Scler. 2011, 17, 1384–1386.
- Allbutt, T.C. On the ophthalmoscopic signs of spinal disease. Lancet 1870, 95, 76–78.
- Erb, W. Ueber das zusammenvorkommen von neuritis optica und myelitis subacuta. Eur. Arch. Psychiatry Clin. Neurosci. 1880, 10, 146–157.
- Seguin, E.C. Art. I.—On the coincidence of optic neuritis and subacute transverse myelitis. J. Nerv. Ment. Dis. 1880, 7, 177–188.
- Marques, A. Da neuromielite ótica: Contribuição clínica e etiológica. Hospital 1943, 24, 49–63.
- Lennon, V.A.; Wingerchuk, D.M.; Kryzer, T.J.; Pittock, S.J.; Lucchinetti, C.F.; Fujihara, K.; Nakashima, I.; Weinshenker, B.G. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet 2004, 364, 2106–2112.
- Lennon, V.A.; Kryzer, T.J.; Pittock, S.J.; Verkman, A.S.; Hinson, S.R. Igg marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med. 2005, 202, 473–477.
- Wingerchuk, D.M.; Lennon, V.A.; Lucchinetti, C.F.; Pittock, S.J.; Weinshenker, B.G. The spectrum of neuromyelitis optica. Lancet Neurol. 2007, 6, 805–815.
- Lana-Peixoto, M.A.; Callegaro, D. The expanded spectrum of neuromyelitis optica: Evidences for a new definition. Arq. Neuro-Psiquiatr. 2012, 70, 807–813.
- Wingerchuk, D.M.; Banwell, B.; Bennett, J.L.; Cabre, P.; Carroll, W.; Chitnis, T.; de Seze, J.; Fujihara, K.; Greenberg, B.; Jacob, A.; et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015, 85, 177–189.
- Waters, P.J.; Pittock, S.J.; Bennett, J.L.; Jarius, S.; Weinshenker, B.G.; Wingerchuk, D.M. Evaluation of aquaporin-4 antibody assays. Clin. Exp. Neuroimmunol. 2014, 5, 290–303.
- Jin, B.J.; Rossi, A.; Verkman, A.S. Model of aquaporin-4 supramolecular assembly in orthogonal arrays based on heterotetrameric association of m1-m23 isoforms. Biophys. J. 2011, 100, 2936–2945.
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 2012, 11, 535–544.
- Verkman, A.S.; Anderson, M.O.; Papadopoulos, M.C. Aquaporins: Important but elusive drug targets. Nat. Rev. Drug Discov. 2014, 13, 259–277.
- Saadoun, S.; Waters, P.; Bell, B.A.; Vincent, A.; Verkman, A.S.; Papadopoulos, M.C. Intra-cerebral injection of neuromyelitis optica immunoglobulin g and human complement produces neuromyelitis optica lesions in mice. Brain J. Neurol. 2010, 133, 349–361.
- Lucchinetti, C.F.; Mandler, R.N.; McGavern, D.; Bruck, W.; Gleich, G.; Ransohoff, R.M.; Trebst, C.; Weinshenker, B.; Wingerchuk, D.; Parisi, J.E.; et al. A role for humoral mechanisms in the pathogenesis of devic’s neuromyelitis optica. Brain J. Neurol. 2002, 125, 1450–1461.
- Jarius, S.; Franciotta, D.; Paul, F.; Ruprecht, K.; Bergamaschi, R.; Rommer, P.S.; Reuss, R.; Probst, C.; Kristoferitsch, W.; Wandinger, K.P.; et al. Cerebrospinal fluid antibodies to aquaporin-4 in neuromyelitis optica and related disorders: Frequency, origin, and diagnostic relevance. J. Neuroinflamm. 2010, 7, 52.
- Chihara, N.; Aranami, T.; Sato, W.; Miyazaki, Y.; Miyake, S.; Okamoto, T.; Ogawa, M.; Toda, T.; Yamamura, T. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc. Natl. Acad. Sci. USA 2011, 108, 3701–3706.
- Shimizu, F.; Schaller, K.L.; Owens, G.P.; Cotleur, A.C.; Kellner, D.; Takeshita, Y.; Obermeier, B.; Kryzer, T.J.; Sano, Y.; Kanda, T.; et al. Glucose-regulated protein 78 autoantibody associates with blood-brain barrier disruption in neuromyelitis optica. Sci. Transl. Med. 2017, 9, eaai9111.
- Shimizu, F.; Nishihara, H.; Kanda, T. Blood-brain barrier dysfunction in immuno-mediated neurological diseases. Immunol. Med. 2018, 41, 120–128.
- Jarius, S.; Wildemann, B.; Paul, F. Neuromyelitis optica: Clinical features, immunopathogenesis and treatment. Clin. Exp. Immunol. 2014, 176, 149–164.
- Weinshenker, B.G.; Wingerchuk, D.M. Neuromyelitis spectrum disorders. Mayo Clin. Proc. 2017, 92, 663–679.
- Hinson, S.R.; Pittock, S.J.; Lucchinetti, C.F.; Roemer, S.F.; Fryer, J.P.; Kryzer, T.J.; Lennon, V.A. Pathogenic potential of igg binding to water channel extracellular domain in neuromyelitis optica. Neurology 2007, 69, 2221–2231.
- Bennett, J.L.; Lam, C.; Kalluri, S.R.; Saikali, P.; Bautista, K.; Dupree, C.; Glogowska, M.; Case, D.; Antel, J.P.; Owens, G.P.; et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann. Neurol. 2009, 66, 617–629.
- Bradl, M.; Misu, T.; Takahashi, T.; Watanabe, M.; Mader, S.; Reindl, M.; Adzemovic, M.; Bauer, J.; Berger, T.; Fujihara, K.; et al. Neuromyelitis optica: Pathogenicity of patient immunoglobulin in vivo. Ann. Neurol. 2009, 66, 630–643.
- Kinoshita, M.; Nakatsuji, Y.; Kimura, T.; Moriya, M.; Takata, K.; Okuno, T.; Kumanogoh, A.; Kajiyama, K.; Yoshikawa, H.; Sakoda, S. Neuromyelitis optica: Passive transfer to rats by human immunoglobulin. Biochem. Biophys. Res. Commun. 2009, 386, 623–627.
- Kinoshita, M.; Nakatsuji, Y.; Kimura, T.; Moriya, M.; Takata, K.; Okuno, T.; Kumanogoh, A.; Kajiyama, K.; Yoshikawa, H.; Sakoda, S. Anti-aquaporin-4 antibody induces astrocytic cytotoxicity in the absence of cns antigen-specific t cells. Biochem. Biophys. Res. Commun. 2010, 394, 205–210.
- Etemadifar, M.; Nasr, Z.; Khalili, B.; Taherioun, M.; Vosoughi, R. Epidemiology of neuromyelitis optica in the world: A systematic review and meta-analysis. Mult. Scler. Int. 2015, 2015, 174720.
- Flanagan, E.P.; Cabre, P.; Weinshenker, B.G.; Sauver, J.S.; Jacobson, D.J.; Majed, M.; Lennon, V.A.; Lucchinetti, C.F.; McKeon, A.; Matiello, M.; et al. Epidemiology of aquaporin-4 autoimmunity and neuromyelitis optica spectrum. Ann. Neurol. 2016, 79, 775–783.
- Rivera, J.F.; Kurtzke, J.F.; Booth, V.A.; Corona, T. Characteristics of devic’s disease (neuromyelitis optica) in mexico. J. Neurol. 2008, 255, 710–715.
- Asgari, N.; Lillevang, S.T.; Skejoe, H.P.; Falah, M.; Stenager, E.; Kyvik, K.O. A population-based study of neuromyelitis optica in caucasians. Neurology 2011, 76, 1589–1595.
- Mori, M.; Kuwabara, S.; Paul, F. Worldwide prevalence of neuromyelitis optica spectrum disorders. J. Neurol. Neurosurg. Psychiatry 2018, 89, 555–556.
- Bukhari, W.; Prain, K.M.; Waters, P.; Woodhall, M.; O’Gorman, C.M.; Clarke, L.; Silvestrini, R.A.; Bundell, C.S.; Abernethy, D.; Bhuta, S.; et al. Incidence and prevalence of nmosd in australia and new zealand. J. Neurol. Neurosurg. Psychiatry 2017, 88, 632–638.
- Kim, S.H.; Mealy, M.A.; Levy, M.; Schmidt, F.; Ruprecht, K.; Paul, F.; Ringelstein, M.; Aktas, O.; Hartung, H.P.; Asgari, N.; et al. Racial differences in neuromyelitis optica spectrum disorder. Neurology 2018, 91, e2089–e2099.
- Cabrera-Gomez, J.A.; Kurtzke, J.F.; Gonzalez-Quevedo, A.; Lara-Rodriguez, R. An epidemiological study of neuromyelitis optica in cuba. J. Neurol. 2009, 256, 35–44.
- Kitley, J.; Leite, M.I.; Nakashima, I.; Waters, P.; McNeillis, B.; Brown, R.; Takai, Y.; Takahashi, T.; Misu, T.; Elsone, L.; et al. Prognostic factors and disease course in aquaporin-4 antibody-positive patients with neuromyelitis optica spectrum disorder from the united kingdom and japan. Brain J. Neurol. 2012, 135, 1834–1849.
- Sepulveda, M.; Armangue, T.; Sola-Valls, N.; Arrambide, G.; Meca-Lallana, J.E.; Oreja-Guevara, C.; Mendibe, M.; Alvarez de Arcaya, A.; Aladro, Y.; Casanova, B.; et al. Neuromyelitis optica spectrum disorders: Comparison according to the phenotype and serostatus. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e225.
- McKeon, A.; Lennon, V.A.; Lotze, T.; Tenenbaum, S.; Ness, J.M.; Rensel, M.; Kuntz, N.L.; Fryer, J.P.; Homburger, H.; Hunter, J.; et al. Cns aquaporin-4 autoimmunity in children. Neurology 2008, 71, 93–100.
- Matiello, M.; Kim, H.J.; Kim, W.; Brum, D.G.; Barreira, A.A.; Kingsbury, D.J.; Plant, G.T.; Adoni, T.; Weinshenker, B.G. Familial neuromyelitis optica. Neurology 2010, 75, 310–315.
- Matsushita, T.; Matsuoka, T.; Isobe, N.; Kawano, Y.; Minohara, M.; Shi, N.; Nishimura, Y.; Ochi, H.; Kira, J. Association of the hla-dpb1*0501 allele with anti-aquaporin-4 antibody positivity in japanese patients with idiopathic central nervous system demyelinating disorders. Tissue Antigens 2009, 73, 171–176.
- Wang, H.; Dai, Y.; Qiu, W.; Zhong, X.; Wu, A.; Wang, Y.; Lu, Z.; Bao, J.; Hu, X. Hla-dpb1 0501 is associated with susceptibility to anti-aquaporin-4 antibodies positive neuromyelitis optica in southern han chinese. J. Neuroimmunol. 2011, 233, 181–184.
- Blanco, Y.; Ercilla-Gonzalez, G.; Llufriu, S.; Casanova-Estruch, B.; Magraner, M.J.; Ramio-Torrenta, L.; Mendibe-Bilbao, M.M.; Ucles-Sanchez, A.J.; Casado-Chocan, J.L.; Lopez de Munain, A.; et al. hla-drb1 typing in caucasians patients with neuromyelitis optica. Rev. De Neurol. 2011, 53, 146–152.
- Pandit, L.; Malli, C.; D’Cunha, A.; Mustafa, S. Human leukocyte antigen association with neuromyelitis optica in a south indian population. Mult. Scler. 2015, 21, 1217–1218.
- Deschamps, R.; Paturel, L.; Jeannin, S.; Chausson, N.; Olindo, S.; Bera, O.; Bellance, R.; Smadja, D.; Cesaire, D.; Cabre, P. Different hla class ii (drb1 and dqb1) alleles determine either susceptibility or resistance to nmo and multiple sclerosis among the french afro-caribbean population. Mult. Scler. 2011, 17, 24–31.
- Zephir, H.; Fajardy, I.; Outteryck, O.; Blanc, F.; Roger, N.; Fleury, M.; Rudolf, G.; Marignier, R.; Vukusic, S.; Confavreux, C.; et al. Is neuromyelitis optica associated with human leukocyte antigen? Mult. Scler. 2009, 15, 571–579.
- Flanagan, E.P.; Weinshenker, B.G.; Krecke, K.N.; Lennon, V.A.; Lucchinetti, C.F.; McKeon, A.; Wingerchuk, D.M.; Shuster, E.A.; Jiao, Y.; Horta, E.S.; et al. Short myelitis lesions in aquaporin-4-igg-positive neuromyelitis optica spectrum disorders. JAMA Neurol. 2015, 72, 81–87.
- Pittock, S.J.; Weinshenker, B.G.; Lucchinetti, C.F.; Wingerchuk, D.M.; Corboy, J.R.; Lennon, V.A. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch. Neurol. 2006, 63, 964–968.
- Matthews, L.; Marasco, R.; Jenkinson, M.; Kuker, W.; Luppe, S.; Leite, M.I.; Giorgio, A.; De Stefano, N.; Robertson, N.; Johansen-Berg, H.; et al. Distinction of seropositive nmo spectrum disorder and ms brain lesion distribution. Neurology 2013, 80, 1330–1337.
- Kim, H.J.; Paul, F.; Lana-Peixoto, M.A.; Tenembaum, S.; Asgari, N.; Palace, J.; Klawiter, E.C.; Sato, D.K.; de Seze, J.; Wuerfel, J.; et al. Mri characteristics of neuromyelitis optica spectrum disorder: An international update. Neurology 2015, 84, 1165–1173.
- Jurynczyk, M.; Tackley, G.; Kong, Y.; Geraldes, R.; Matthews, L.; Woodhall, M.; Waters, P.; Kuker, W.; Craner, M.; Weir, A.; et al. Brain lesion distribution criteria distinguish ms from aqp4-antibody nmosd and mog-antibody disease. J. Neurol. Neurosurg. Psychiatry 2017, 88, 132–136.
- Kira, J.I. Unexpected exacerbations following initiation of disease-modifying drugs in neuromyelitis optica spectrum disorder: Which factor is responsible, anti-aquaporin 4 antibodies, b cells, th1 cells, th2 cells, th17 cells, or others? Mult. Scler. 2017, 23, 1300–1302.
- Kleiter, I.; Gahlen, A.; Borisow, N.; Fischer, K.; Wernecke, K.D.; Wegner, B.; Hellwig, K.; Pache, F.; Ruprecht, K.; Havla, J.; et al. Neuromyelitis optica: Evaluation of 871 attacks and 1,153 treatment courses. Ann. Neurol. 2016, 79, 206–216.
- Weinshenker, B.G. What is the optimal sequence of rescue treatments for attacks of neuromyelitis optica spectrum disorder? Ann. Neurol. 2016, 79, 204–205.
- Wingerchuk, D.M.; Weinshenker, B.G. Neuromyelitis optica. Curr. Treat. Options Neurol. 2008, 10, 55–66.
- Kim, S.H.; Hyun, J.W.; Kim, H.J. Individualized b cell-targeting therapy for neuromyelitis optica spectrum disorder. Neurochem. Int. 2018, in press.
- Paul, F.; Murphy, O.; Pardo, S.; Levy, M. Investigational drugs in development to prevent neuromyelitis optica relapses. Expert Opin. Investig. Drugs 2018, 27, 265–271.
- Kelly, R.J.; Hochsmann, B.; Szer, J.; Kulasekararaj, A.; de Guibert, S.; Roth, A.; Weitz, I.C.; Armstrong, E.; Risitano, A.M.; Patriquin, C.J.; et al. Eculizumab in pregnant patients with paroxysmal nocturnal hemoglobinuria. N. Engl. J. Med. 2015, 373, 1032–1039.
- McNamara, L.A.; Topaz, N.; Wang, X.; Hariri, S.; Fox, L.; MacNeil, J.R. High risk for invasive meningococcal disease among patients receiving eculizumab (soliris) despite receipt of meningococcal vaccine. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2017, 17, 2481–2484.
- Traboulsee, A.; Greenberg, B.; Bennett, J.L.; Szczechowiski, L.; Fox, E.; Shkrobot, S.; Yamamura, T.; Terada, Y.; Kawata, Y.; Melia, A.; et al. A double-blind placebo-controlled study of satralizumab (sa 237), a recycling anti-il-6 receptor monoclonal antibody, as monotherapy for patients witn neuromyelitis optica spectrum disorder (nmosd). In Proceedings of the ECTRIMS, Berlin, Germany, 10–12 Octorber 2018. Poster 1278.
- Steinman, L.; Bar-Or, A.; Behne, J.M.; Benitez-Ribas, D.; Chin, P.S.; Clare-Salzler, M.; Healey, D.; Kim, J.I.; Kranz, D.M.; Lutterotti, A.; et al. Restoring immune tolerance in neuromyelitis optica: Part i. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e276.
More
Information
Subjects:
Medicine, General & Internal
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
813
Revisions:
2 times
(View History)
Update Date:
01 Sep 2021
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No