Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Osman El‐Maarri | + 1578 word(s) | 1578 | 2021-07-30 09:54:29 | | | |
| 2 | Ron Wang | + 571 word(s) | 2149 | 2021-08-16 04:07:05 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
El‐Maarri, O. Assisted Reproductive Techniques (ART). Encyclopedia. Available online: https://encyclopedia.pub/entry/13146 (accessed on 24 July 2026).
El‐Maarri O. Assisted Reproductive Techniques (ART). Encyclopedia. Available at: https://encyclopedia.pub/entry/13146. Accessed July 24, 2026.
El‐Maarri, Osman. "Assisted Reproductive Techniques (ART)" Encyclopedia, https://encyclopedia.pub/entry/13146 (accessed July 24, 2026).
El‐Maarri, O. (2021, August 13). Assisted Reproductive Techniques (ART). In Encyclopedia. https://encyclopedia.pub/entry/13146
El‐Maarri, Osman. "Assisted Reproductive Techniques (ART)." Encyclopedia. Web. 13 August, 2021.
Copy Citation
To increase the efficiency of assisted reproductive techniques (ART), molecular studies have been performed to identify the best predictive biomarkers for selecting the most suitable germ cells for fertilization and the best embryo for intra-uterine transfer.
ART
ICSI
cumulus cell
mRNA expression profiling
CpG methylation profiling
oocyte competence
epigenetic biomarker
FSH
pathways analysis
upstream regulators
1. Introduction
Infertility is a global concern affecting an estimated 10% of reproductive-age couples [1]. When sperm motility, morphology, or counts are affected, intracytoplasmic sperm injection (ICSI) must be used [2]. Despite technological advances, successful live birth rates are far from 100%. Several markers/parameters are investigated to monitor the entire process and to select the best embryos for intra-uterine transfer to increase the rate of live births. These potential markers are linked to the biological process of follicular and oocyte development, maturation, and ovulation and include a variety of types: (A) endometrium wall thickness and volume [3], (B) hormone levels [4], (C) follicle number and size [5], (D) gene expression in granulosa [6] or mural granulosa cells [7], (E) gene expression in cumulus cells [6][7][8][9][10][11][12][13][14][15][16][17][18][19][20][21][22][23][24][25][26][27], (F) microRNA expression in cumulus cells [28], (G) gene expression in follicular fluid cells [29], (H) zona pellucida morphology parameters [30], (I) analysis of polar body chromosome number and structure [31], and (J) morphological appearance of the oocyte [32].
Of the above-mentioned approaches, gene expression analysis (categories D to G) offers a promising methodology for monitoring oocyte development due to its relative simplicity. However, different studies have failed to identify universal markers linked with good developmental potentials (Supplementary Table S1). This is possibly due to the heterogeneity of samples and to differences in their preparation because sample sources could be mural cells, cumulus cells (inner or outer), granulosa cells (precursor of mural and cumulus cells), or follicular fluid derived cells. The heterogeneity of samples in biological developmental stage and purity of sample preparation, further contributes to the inconsistency of results. Moreover, the degree of maturity of the retrieved oocyte has been found to be associated with a specific expression signature that could further cause discrepancies when comparing results between different studies [33][34]. Likewise, differences in methodology also contribute to the variability of results, as a number of studies have applied broad genomic-based approaches to identify genome-wide expression using microarrays or RNA-seq, while others have focused on the expression of a limited number of genes related to specific pathway(s) that were shown to play important roles in oogenesis. Additionally, different treatment protocols further increase the variability of results.
Several studies performed controlled experiments to study the effect of parameters/factors that could introduce systematic bias into the results. Indeed, age [35], stimulated in vitro fertilization cycles rather than natural cycles [36], differences in treatment protocols, as well as medications used during ovarian stimulation [37][38][39] were seen to have an effect. Additionally, a patient’s genetic constituents also affect the clinical output and success of treatment. For example, the influence of polymorphism in the follicle stimulating hormone receptor (FSHR) and the luteinizing hormone (LH) beta subunit [40] and alternative skipping of exon 2 or 3 in the FSHR [41] have been reported to affect the ovarian responses to the FSH.
2. Current Insights
One of the aims of improving the clinical results of ICSI cycles is to predict the developmental potential of the transferred embryo, thus choosing the best for transfer and leading to less psychological stress for the affected couples and cost reductions for the health system. The use of biomarkers in cumulus cells surrounding the oocyte to predict the clinical outcome is a possible non-invasive approach. To this end, we performed molecular profiling, including, for the first time, simultaneous RNA and DNA methylation profiling on outer cumulus cells retrieved from ICSI cycles.
In order to obtain informative biomarker(s) without false positive influence of interfering factors, we determined, using literature-based knowledge and our experimentally obtained data, conditions that could potentially influence the results (i.e., choice of biomarkers). The main factors influencing the expression and methylation markers were inter-individual differences and differences in treatment protocol: short vs. long.
The first could be a reflection on the genetic setup of individuals and their response to treatment. The inter-individual differences could be clearly observed (Figure 1A) in the unsupervised PCA samples and heatmap where the samples derived from the same individual, even for two different stimulations using the same protocol, were interconnected and clustered close together. This could be explained by genetic polymorphisms affecting general expression patterns or a response to hormone stimulation treatments. It is documented that genetic variations affect the global gene expression patterns [42][43] and that any response to induced hormonal treatment could be modulated by polymorphisms in the FSH receptor [40][44]. Therefore, an influence of the individual genetic make-up on the treatment outcome cannot be avoided. However, it could be predicted from previously known polymorphisms, where the treatment regimens had been precisely adopted for individual genetic setup. Unfortunately, in this study, we could not investigate whether the individual-specific SNPs were directly associated with cumulus expression profile, as patient DNA (from peripheral blood) was unavailable for this study. The second observed influence was the hormonal stimulation protocol, as the differences in the unsupervised classification of samples were clearly influenced by the protocol used, short or long, which reflected the use of different stimulating/treatment regimens (Supplementary Figure S5A). Therefore, we stratified the samples according to treatment protocol but could not do the same for the genomic/polymorphism because such information was not available.
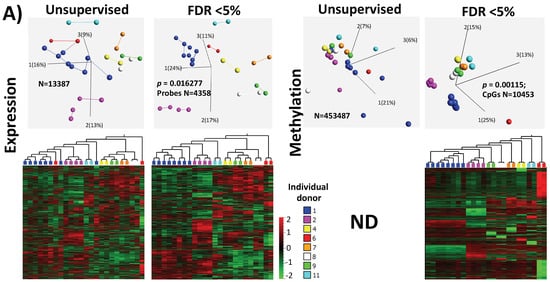
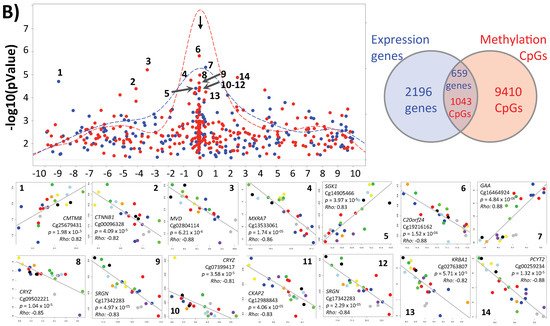
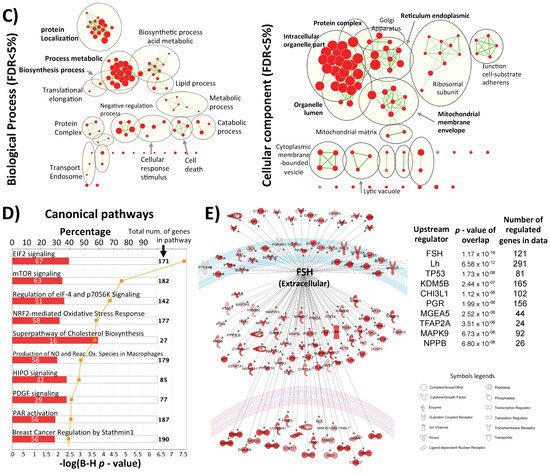

Figure 1. Inter-individual differences in expression and methylation. (A) 3D-PCA (upper part) and heatmaps (lower part) representing expression and CpG methylation profiling of 24 individual cumulus cells derived from eight different women. Left and right parts represent expression and methylation, respectively, unsupervised and ANOVA for multiple comparisons at FDR < 5% analysis are shown. (B) Correlation between expression and methylation. Right upper part shows Venn diagram of intersection/overlap of DEG and DMC; in total 659 genes overlapped with 1043 investigated CpGs. The correlation of the overlaps is shown at the left upper part as −log10 (p-values) vs. the relative position to the transcription start site. In addition, the representative density/fitting curve is shown as a dotted line (red or blue represent negative and positive correlation, respectively). The lower panel shows correlation graphs of the highest 14 correlations (three positive and 11 negative) (The color of the dots represents the individual treatment, and only transferred samples were used in this analysis). (C) Ontology analysis of the group of genes showing strong inter-individual differences (ANOVA analysis at FDR < 0.5); both biological process and cellular component are shown. The vertical arrow indicates the TSS; others arrows are linking to the dots with the number(s). (D) Top-10 affected canonical pathways as determined by IPA. The lower x-axis represents the −log(B–H p-value) for enrichment, while the upper x-axis represents the percentages of variable genes in a given pathway. (E) Top-10 upstream regulators (predicted by IPA) are listed with the overlapping p-values and numbers of regulated genes in the data; FSH regulated proteins are represented with symbols reflecting their functions and in their subcellular localization. (F) The top categories in disease and biofunction (predicted by IPA). The top-10 significant subcategories according to p-values are listed below together with the p-values.
We identified individual single markers that were differentially expressed where NME6 and ASAP1 were under expressed in positive samples (i.e., continuing pregnancy). NME6 is the nucleoside diphosphate kinase 6, which specializes in the phosphorylation of nucleotide diphosphate, which regulates on cell growth. NME6 is ubiquitously expressed at low levels in most human tissues but is abundant in the kidney, prostate, ovary, intestine, and spleen [45]. It has also been found to be particularly abundant in zebrafish ovaries [46]. To date, it has not been experimentally proven whether NME6 is active in human cells. However, it seems to be localized in the mitochondria [47], suggesting a possible role in signaling by contributing to nucleoside metabolism and the ratio of different forms. The latter could itself regulate the growth of cumulus cells around the oocytes and probably the oocyte itself. ASAP1 stands for ArfGAP with SH3 Domain, Ankyrin Repeat and PH Domain 1. It plays a role in shaping the actin cytoskeleton and induces proliferation [48][49]. It is also overexpressed in many tumors and could trigger cellular movement and the ability of cells to move and thus promote metastasis [50][51]. As the process of oocyte maturation and ovulation necessitates cellular remodeling surrounding the oocyte it is not surprising that ASAP1 is considered one of the players indicating a proper maturation stage. However, we also saw a correlation of these two markers with endometrium size in the positive samples (Supplementary Figure S6; Supplementary Table S3); therefore, it is questionable whether this is an effect of endometrium development with an effect on implantation that was also detected in cumulus cells or is a marker for oocyte development. It could be that endometrium development is the main determining factor to be considered and that it overwrites any oocyte factor. Future studies that group large samples based on endometrium size should be performed to determine if these two markers are endometrium or cumulus cell specific.
When comparing our gene marker results to previous results obtained from cumulus cell global expression, very little overlap could be identified (p < 0.05) (Supplementary Figure S7 and Supplementary Table S1). Assidi et al. (2011) [15] identified seven markers as the top differentially expressed between positive and negative samples (NRP1, UBQLN1, PSMD6, DPP8, HIST1H4C, CALM1, TOM1), most of which were reconfirmed by a second study by Assidi et al. (2015) [21], except for HIST1H4C and TOM1. Feuerstein et al. (2012) [17] identified three markers (PLIN2, RGS2, ANG), while Xu et al. (2015) [23] determined nine that were confirmed by RT-PCR (BAI2, HDAC1, ELOVL5, ITGA6, RAD51, VCAN, PTGS2, POU5F1, VAMP7) and Demiray et al. (2019) [26] identified five markers (ZFP57, ARHGEF4, SOX8, FLJ39502, CNIH3) according to clinical pregnancy and >2-fold change. Here, two observations must be highlighted: (1) among the above-mentioned top markers, there was no single marker overlap between two research sites, and (2) Assidi et al. clearly confirmed their initial study. To explain this, we proposed that laboratory-specific factors may affect results, by making single-gene markers non-transferable from one site to another. This could be related to (1) the cellular heterogeneity of isolated materials, (2) specific ethnicity of patient cohorts and genetic make-up, (3) the stimulation and triggering protocol of hormone treatment, (4) experimental procedures of isolating the tested materials, (5) data generation methods (i.e., RNA seq, microarrays or even RT-PCR), and (6) genetic heterogeneity of the patients. The three overlaps from the above-mentioned potential markers within our data are TOM1 (logFC = −0.28; p = 0.00157), HDAC1 (logFC = 0.35; p = 0.00239), and PTGS2 (logFC = −0.61; p = 0.0298).
Although no significant overlap in the single-marker genes from cumulus cells across different studies was observed, the use of gene expression as a biomarker for embryo quality was also questioned by others [52]. However, the increasing trend of using pathway analysis showed common affected pathways. Indeed, IPA and KEGG pathway analyses are able to show which pathways are controlled by differentially expressed genes and which distinguish positive from negative samples. In this respect, our data are in accordance with the general trend of considering apoptotic markers as predictive markers [15][26][53]. Our results also showed similarly affected pathways, such as cell death and survival in the category of bio function (for inter-individual differences see Figure 1C,D,F; for differences between positive and negative samples see Figure 2D,E,G).
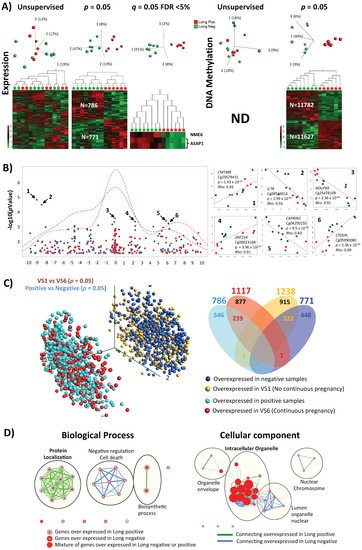
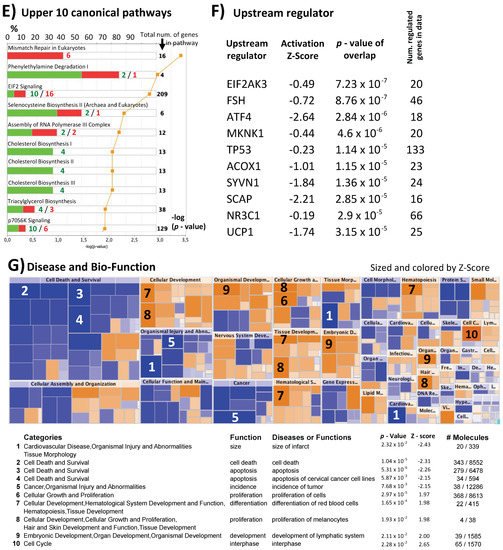
Figure 2. Differences in expression and CpG methylation profiles between pregnant positive and pregnant negative samples of the long protocol. (A) 3D-PCA plots and heatmaps of expression and CpGs methylation profiling. Result of unsupervised and significance at p = 0.05 and FDR < 5%, when applicable, are shown. (B) Correlation between differentially expression genes and differentially methylated CpGs. The correlation of the two data overlaps is shown on the left as −log10 (p-values) vs. the relative position to transcription start site (TSS = 0; every unit is 1 Kb), representative density/fitting curve is also shown in dotted line (red or blue represents negative or positive correlation, respectively). The right panel shows individual data for the highest six correlations (three positive and three negative correlations) (the color of the dots represents the pregnancy test result with red (positive) and green (negative)). (C) Comparison between results of successive analyses of two treatments of the same individual (VS1 negative vs. VS6 positive; shown in Figure 2) on one side and comparison between all pregnancy negative and pregnancy positive samples on the other (part A above). The left part represents the 3D variables of the PCA expression data (part A above), where the overlap of the overexpressed and the underexpressed genes between the two comparisons is in a near-complete phase. The right part represents a Venn diagram showing the overlap of the differentially expressed genes for the two comparisons in question. (D) Ontology analysis of the genes with difference in expression at p = 0.05 for biological process and cellular component are shown. IPA analysis (at p < 0.05%) for top-10 affected (E) canonical pathway, (F) upstream regulators (details of FSH-affected targets are shown on the left), and (G) diseases and biofunction.
References
- Inhorn, M.C.; Patrizio, P. Infertility around the globe: New thinking on gender, reproductive technologies and global movements in the 21st century. Hum. Reprod. Update 2015, 21, 411–426.
- Palemro, G.; Joris, H.; Devroey, P.; Van Steirteghem, A.C. Pregnancies after intercytoplasmic injection of single spermatozoon into an oocyte. Lancet 1992, 340, 17–18.
- Yuan, X.; Saravelos, S.H.; Wang, Q.; Xu, Y.; Li, T.C.; Zhou, C. Endometrial thickness as a predictor of pregnancy outcomes in 10787 fresh IVF-ICSI cycles. Reprod. Biomed. Online 2016, 33, 197–205.
- Ioannidis, G.; Sacks, G.; Reddy, N.; Seyani, L.; Margara, R.; Lavery, S.; Trew, G. Day 14 maternal serum progesterone levels predict pregnancy outcome in IVF/ICSI treatment cycles: A prospective study. Hum. Reprod. 2005, 20, 741–746.
- Abbara, A.; Vuong, L.N.; Ho, V.N.A.; Clarke, S.A.; Jeffers, L.; Comninos, A.N.; Salim, R.; Ho, T.M.; Kelsey, T.W.; Trew, G.H. Follicle Size on Day of Trigger Most Likely to Yield a Mature Oocyte. Front. Endocrinol. 2018, 9, 193.
- Maman, E.; Yung, Y.; Kedem, A.; Yerushalmi, G.M.; Konopnicki, S.; Cohen, B.; Dor, J.; Hourvitz, A. High expression of luteinizing hormone receptors messenger RNA by human cumulus granulosa cells is in correlation with decreased fertilization. Fertil. Steril. 2012, 97, 592–598.
- Grondahl, M.L.; Andersen, C.Y.; Bogstad, J.; Borgbo, T.; Boujida, V.H.; Borup, R. Specific genes are selectively expressed between cumulus and granulosa cells from individual human pre-ovulatory follicles. Mol. Hum. Reprod. 2012, 18, 572–584.
- McKenzie, L.J.; Pangas, S.A.; Carson, S.A.; Kovanci, E.; Cisneros, P.; Buster, J.E.; Amato, P.; Matzuk, M.M. Human cumulus granulosa cell gene expression: A predictor of fertilization and embryo selection in women undergoing IVF. Hum. Reprod. 2004, 19, 2869–2874.
- Hasegawa, J.; Yanaihara, A.; Iwasaki, S.; Mitsukawa, K.; Negishi, M.; Okai, T. Reduction of connexin 43 in human cumulus cells yields good embryo competence during ICSI. J. Assist. Reprod. Genet. 2007, 24, 463–466.
- Anderson, R.A.; Sciorio, R.; Kinnell, H.; Bayne, R.A.; Thong, K.J.; de Sousa, P.A.; Pickering, S. Cumulus gene expression as a predictor of human oocyte fertilisation, embryo development and competence to establish a pregnancy. Reproduction 2009, 138, 629–637.
- Haouzi, D.; Assou, S.; Mahmoud, K.; Hedon, B.; De Vos, J.; Dewailly, D.; Hamamah, S. LH/hCGR gene expression in human cumulus cells is linked to the expression of the extracellular matrix modifying gene TNFAIP6 and to serum estradiol levels on day of hCG administration. Hum. Reprod. 2009, 24, 2868–2878.
- Wang, H.X.; Tong, D.; El-Gehani, F.; Tekpetey, F.R.; Kidder, G.M. Connexin expression and gap junctional coupling in human cumulus cells: Contribution to embryo quality. J. Cell. Mol. Med. 2009, 13, 972–984.
- Adriaenssens, T.; Wathlet, S.; Segers, I.; Verheyen, G.; De Vos, A.; Van der Elst, J.; Coucke, W.; Devroey, P.; Smitz, J. Cumulus cell gene expression is associated with oocyte developmental quality and influenced by patient and treatment characteristics. Hum. Reprod. 2010, 25, 1259–1270.
- Koks, S.; Velthut, A.; Sarapik, A.; Altmae, S.; Reinmaa, E.; Schalkwyk, L.C.; Fernandes, C.; Lad, H.V.; Soomets, U.; Jaakma, U.; et al. The differential transcriptome and ontology profiles of floating and cumulus granulosa cells in stimulated human antral follicles. Mol. Hum. Reprod. 2010, 16, 229–240.
- Assidi, M.; Montag, M.; Van der Ven, K.; Sirard, M.A. Biomarkers of human oocyte developmental competence expressed in cumulus cells before ICSI: A preliminary study. J. Assist. Reprod. Genet. 2011, 28, 173–188.
- Wathlet, S.; Adriaenssens, T.; Segers, I.; Verheyen, G.; Van de Velde, H.; Coucke, W.; Ron El, R.; Devroey, P.; Smitz, J. Cumulus cell gene expression predicts better cleavage-stage embryo or blastocyst development and pregnancy for ICSI patients. Hum. Reprod. 2011, 26, 1035–1051.
- Feuerstein, P.; Puard, V.; Chevalier, C.; Teusan, R.; Cadoret, V.; Guerif, F.; Houlgatte, R.; Royere, D. Genomic assessment of human cumulus cell marker genes as predictors of oocyte developmental competence: Impact of various experimental factors. PLoS ONE 2012, 7, e40449.
- Varras, M.; Griva, T.; Kalles, V.; Akrivis, C.; Paparisteidis, N. Markers of stem cells in human ovarian granulosa cells: Is there a clinical significance in ART? J. Ovarian Res. 2012, 5, 36.
- Wathlet, S.; Adriaenssens, T.; Segers, I.; Verheyen, G.; Van Landuyt, L.; Coucke, W.; Devroey, P.; Smitz, J. Pregnancy prediction in single embryo transfer cycles after ICSI using QPCR: Validation in oocytes from the same cohort. PLoS ONE 2013, 8, e54226.
- Li, Y.; Li, R.-Q.; Song-Bang Ou, S.-B.; Zhang, N.-F.; Ren, L.; Wei, L.-N.; Zhang, Q.; Yang, D. Increased GDF9 and BMP15 mRNA levels in cumulus granulosa cells correlate with oocyte maturation, fertilization, and embryo quality in humans. Reprod. Biol. Endocrinol. RB&E 2014, 12, 1–9.
- Assidi, M.; Montag, M.; Sirard, M.A. Use of Both Cumulus Cells’ Transcriptomic Markers and Zona Pellucida Birefringence to Select Developmentally Competent Oocytes in Human Assisted Reproductive Technologies. BMC Genom. 2015, 16, 9.
- Burnik Papler, T.; Vrtacnik Bokal, E.; Maver, A.; Lovrecic, L. Specific gene expression differences in cumulus cells as potential biomarkers of pregnancy. Reprod. Biomed. Online 2015, 30, 426–433.
- Xu, X.; Chen, D.; Zhang, Z.; Wei, Z.; Cao, Y. Molecular signature in human cumulus cells related to embryonic developmental potential. Reprod. Sci. 2015, 22, 173–180.
- Artini, P.G.; Tatone, C.; Sperduti, S.; D’Aurora, M.; Franchi, S.; Di Emidio, G.; Ciriminna, R.; Vento, M.; di Pietro, C.; Stuppia, L.; et al. Cumulus cells surrounding oocytes with high developmental competence exhibit down-regulation of phosphoinositol 1,3 kinase/protein kinase B (PI3K/AKT) signalling genes involved in proliferation and survival. Hum. Reprod. 2017, 32, 2474–2484.
- Green, K.A.; Franasiak, J.M.; Werner, M.D.; Tao, X.; Landis, J.N.; Scott, R.T., Jr.; Treff, N.R. Cumulus cell transcriptome profiling is not predictive of live birth after in vitro fertilization: A paired analysis of euploid sibling blastocysts. Fertil. Steril. 2018, 109, 460–466.
- Demiray, S.B.; Goker, E.N.T.; Tavmergen, E.; Yilmaz, O.; Calimlioglu, N.; Soykam, H.O.; Oktem, G.; Sezerman, U. Differential gene expression analysis of human cumulus cells. Clin. Exp. Reprod. Med. 2019, 46, 76–86.
- Faramarzi, A.; Khalili, M.A.; Jahromi, M.G. Is there any correlation between apoptotic genes expression in cumulus cells with embryo morphokinetics? Mol. Biol. Rep. 2019, 46, 3663–3670.
- Karakaya, C.; Guzeloglu-Kayisli, O.; Uyar, A.; Kallen, A.N.; Babayev, E.; Bozkurt, N.; Unsal, E.; Karabacak, O.; Seli, E. Poor ovarian response in women undergoing in vitro fertilization is associated with altered microRNA expression in cumulus cells. Fertil. Steril. 2015, 103, 1469–1476.
- Hamel, M.; Dufort, I.; Robert, C.; Gravel, C.; Leveille, M.C.; Leader, A.; Sirard, M. Identification of differentially expressed markers in human follicular cells associated with competent oocytes. Hum. Reprod. 2008, 23, 1118–1127.
- Ebner, T.; Balaban, B.; Moser, M.; Shebl, O.; Urman, B.; Ata, B.; Tews, G. Automatic user-independent zona pellucida imaging at the oocyte stage allows for the prediction of preimplantation development. Fertil. Steril. 2010, 94, 913–920.
- Verpoest, W.; Staessen, C.; Bossuyt, P.M.; Goossens, V.; Altarescu, G.; Bonduelle, M.; Devesa, M.; Eldar-Geva, T.; Gianaroli, L.; Griesinger, G.; et al. Preimplantation genetic testing for aneuploidy by microarray analysis of polar bodies in advanced maternal age: A randomized clinical trial. Hum. Reprod. 2018, 33, 1767–1776.
- Zanetti, B.F.; Braga, D.; Setti, S.; Figueira, D.C.S.; Iaconelli, A.; Borges, E. Is perivitelline space morphology of the oocyte associated with pregnancy outcome in intracytoplasmic sperm injection cycles? Eur. J. Obstet. Gynecol. Reprod. Biol. 2018, 231, 225–229.
- Ouandaogo, Z.G.; Frydman, N.; Hesters, L.; Assou, S.; Haouzi, D.; Dechaud, H.; Frydman, R.; Hamamah, S. Differences in transcriptomic profiles of human cumulus cells isolated from oocytes at GV, MI and MII stages after in vivo and in vitro oocyte maturation. Hum. Reprod. 2012, 27, 2438–2447.
- Ouandaogo, Z.G.; Haouzi, D.; Assou, S.; Dechaud, H.; Kadoch, I.J.; De Vos, J.; Hamamah, S. Human cumulus cells molecular signature in relation to oocyte nuclear maturity stage. PLoS ONE 2011, 6, e27179.
- Al-Edani, T.; Assou, S.; Ferrieres, A.; Bringer Deutsch, S.; Gala, A.; Lecellier, C.H.; Ait-Ahmed, O.; Hamamah, S. Female aging alters expression of human cumulus cells genes that are essential for oocyte quality. BioMed Res. Int. 2014, 2014, 964614.
- Papler, T.B.; Bokal, E.V.; Tacer, K.F.; Juvan, P.; Virant Klun, I.; Devjak, R. Differences in cumulus cells gene expression between modified natural and stimulated in vitro fertilization cycles. J. Assist. Reprod. Genet. 2014, 31, 79–88.
- Borgbo, T.; Povlsen, B.B.; Andersen, C.Y.; Borup, R.; Humaidan, P.; Grondahl, M.L. Comparison of gene expression profiles in granulosa and cumulus cells after ovulation induction with either human chorionic gonadotropin or a gonadotropin-releasing hormone agonist trigger. Fertil. Steril. 2013, 100, 994–1001.
- Gatta, V.; Tatone, C.; Ciriminna, R.; Vento, M.; Franchi, S.; d’Aurora, M.; Sperduti, S.; Cela, V.; Borzi, P.; Palermo, R.; et al. Gene expression profiles of cumulus cells obtained from women treated with recombinant human luteinizing hormone + recombinant human follicle-stimulating hormone or highly purified human menopausal gonadotropin versus recombinant human follicle-stimulating hormone alone. Fertil. Steril. 2013, 99, 2000–2008.
- Gurgan, T.; Montjean, D.; Demirol, A.; Menezo, Y.J. Sequential (hFSH + recFSH) vs homogenous (hFSH or recFSH alone) stimulation: Clinical and biochemical (cumulus cell gene expression) aspects. J. Assist. Reprod. Genet. 2014, 31, 657–665.
- Bosco, L.; Ruvolo, G.; Luparello, C.; Ferrari, S.; Valerio, D.; Santi, D.; Piomboni, P.; Sarcina, E.; Lispi, M.; Roccheri, M.C. Gene Expression and Apoptosis Levels in Cumulus Cells of Patients with Polymorphisms of FSHR and LHB Undergoing in Vitro Fertilization Program. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 43, 2391–2404.
- Karakaya, C.; Guzeloglu-Kayisli, O.; Hobbs, R.J.; Gerasimova, T.; Uyar, A.; Erdem, M.; Oktem, M.; Erdem, A.; Gumuslu, S.; Ercan, D.; et al. Follicle-stimulating hormone receptor (FSHR) alternative skipping of exon 2 or 3 affects ovarian response to FSH. Mol. Hum. Reprod. 2014, 20, 630–643.
- Ackermann, M.; Sikora-Wohlfeld, W.; Beyer, A. Impact of natural genetic variation on gene expression dynamics. PLoS Genet. 2013, 9, e1003514.
- Consortium, G.T.; Laboratory, D.A. Genetic effects on gene expression across human tissues. Nature 2017, 550, 204–213.
- Wunsch, A.; Sonntag, B.; Simoni, M. Polymorphism of the FSH receptor and ovarian response to FSH. Ann. Endocrinol. 2007, 68, 160–166.
- Mehus, J.G.; Deloukas, P.; Lambeth, D.O. NME6: A new member of the nm23/nucleoside diphosphate kinase gene family located on human chromosome 3p21.3. Hum. Genet. 1999, 104, 454–459.
- Desvignes, T.; Fauvel, C.; Bobe, J. The NME gene family in zebrafish oogenesis and early development. Naunyn Schmiedeberg’s Arch. Pharmacol. 2011, 384, 439–449.
- Bosnar, M.H.; Radić, M.; Ačkar, L.; Proust, B.; Ćetković, H. Subcellular Localization and Expression of Nme6, a Member of The Nme/Nm23/Ndpk Family. Hum. Tumour Cells. 2018, 3, 122.
- Gasilina, A.; Vitali, T.; Luo, R.; Jian, X.; Randazzo, P.A. The ArfGAP ASAP1 Controls Actin Stress Fiber Organization via Its N-BAR Domain. iScience 2019, 22, 166–180.
- Luo, R.; Reed, C.E.; Sload, J.A.; Wordeman, L.; Randazzo, P.A.; Chen, P.W. Arf GAPs and molecular motors. Small GTPases 2019, 10, 196–209.
- Zhang, T.; Zhao, G.; Yang, C.; Dong, P.; Watari, H.; Zeng, L.; Pfeffer, L.M.; Yue, J. Lentiviral vector mediated-ASAP1 expression promotes epithelial to mesenchymal transition in ovarian cancer cells. Oncol. Lett. 2018, 15, 4432–4438.
- Muller, T.; Stein, U.; Poletti, A.; Garzia, L.; Rothley, M.; Plaumann, D.; Thiele, W.; Bauer, M.; Galasso, A.; Schlag, P.; et al. ASAP1 promotes tumor cell motility and invasiveness, stimulates metastasis formation in vivo, and correlates with poor survival in colorectal cancer patients. Oncogene 2010, 29, 2393–2403.
- Kahraman, S.; Cetinkaya, C.P.; Cetinkaya, M.; Tufekci, M.A.; Ekmekci, C.G.; Montag, M. Is there a correlation between follicle size and gene expression in cumulus cells and is gene expression an indicator of embryo development? Reprod. Biol. Endocrinol. RB&E 2018, 16, 69.
- O’Shea, L.C.; Mehta, J.; Lonergan, P.; Hensey, C.; Fair, T. Developmental competence in oocytes and cumulus cells: Candidate genes and networks. Syst. Biol. Reprod. Med. 2012, 58, 88–101.
More
Information
Subjects:
Genetics & Heredity
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
2 times
(View History)
Update Date:
16 Aug 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No