 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yurii Borovikov | + 1071 word(s) | 1071 | 2020-07-09 08:20:36 | | | |
| 2 | Camila Xu | Meta information modification | 1071 | 2020-07-13 07:54:00 | | | | |
| 3 | Camila Xu | -5 word(s) | 1066 | 2020-10-27 09:23:31 | | |
Video Upload Options
Substitution of Glu173 for Ala in Tpm3.12 (E173A) is associated with congenital muscle weakness. It was found that this mutation increases myofilament Ca2+-sensitivity and inhibits in vitro actin-activated ATPase activity of myosin subfragment-1 at high Ca2+. In order to determine the critical conformational changes in myosin, actin and tropomyosin caused by the mutation, we used the polarized fluorimetry technique. We observed changes in the spatial arrangement of actin monomers and myosin heads, and in the position of the mutant tropomyosin on the thin filaments in muscle fibres at various ATPase cycle stages. At low Ca2+ the E173A mutant tropomyosin shifts abnormally towards the inner domains of actin at all stages of the cycle. The number of switched-on actin monomers and strong-binding myosin heads increases even at relaxation. Contrarily, at high Ca2+ the amount of the myosin heads strongly bound with F-actin slightly decreases. The changes in the balance of the strongly bound myosin heads in the ATPase cycle may underlie the occurrence of muscle weakness. W7, an inhibitor of troponin Ca2+-sensitivity, restores the number of strong-binding myosin heads at high Ca2+ and inhibits it at relaxation, suggesting the possibility of using Ca2+-desensitizers to reduce the damaging effect of the E173A mutation on muscle fibre contractility.
1. Introduction
Contraction of skeletal muscle is regulated through the thin filaments, which contain actin, tropomyosin and troponin [1]. When the intracellular concentration of calcium changes, tropomyosin associated with actin and troponin shifts on the surface of the actin filament, opening or closing the sites for binding of the myosin heads on actin. The electrostatic nature of the actin-tropomyosin interaction and flexibility of actin and tropomyosin [2][3] can explain the dynamic displacement of tropomyosin relative to the outer and inner actin domains (between the blocked, closed and open positions) during contraction [2][4][5][6][7]. The change in the position of the tropomyosin strands relative to the actin inner domains is due to the difference between tropomyosin and F-actin in their bending flexibility (therefore, variation in the persistence lengths of these proteins [4][8]), which presumably causes an azimuthal shift of the tropomyosin strands. When Ca2+ binds to troponin-C, some actin monomers change their conformation to the switched-on state, and the persistence length of the actin filament decreases. At the same time, the tropomyosin persistence length increases, and tropomyosin moves towards the actin inner domains, partly exposing the myosin-binding site (closed position). At low Ca2+, troponin-I interacts with actin [9], switching thin filaments off, which leads to spatial rearrangement and an increase in the persistence length of the actin filament. At the same time, the tropomyosin persistence length decreases and tropomyosin is delayed in a position close to the outer actin domains (the blocked position). In this state of the thin filament (the “off” state), the strong binding of myosin with actin is inhibited. When the myosin heads are strongly bound to the F-actin filament, the actin monomers are switched-on, the persistence length of the actin filament decreases, and that of tropomyosin increases. In this state (the “on” state), the tropomyosin strands completely expose the binding sites of F-actin to myosin and, therefore, initiate muscle contraction. It was also found that tropomyosin can bind to the myosin head, regulating the binding of the latter to actin [10]. Consequently, tropomyosin, which is associated with both actin and troponin, is able to bind the myosin head and is also a central link in the regulation of actin-myosin interaction.
In skeletal muscle there are three main tropomyosin isoforms which are encoded by the TPM1, TPM2 and TPM3 genes [11]. Mutations give rise to a wide spectrum of clinically, histologically and genetically variable neuromuscular and cardiac disorders [12]. The numerous point mutations in TPM3 gene were found in patients with such congenital pathologies as nemaline myopathy, distal arthrogryposis, congenital muscle fibre type disproportion, cap-myopathy [13]. The E173A mutation in Tpm3.12 encoded by the TPM3 gene was detected in a 7-year-old boy with hypotonia, feeding difficulties, motor delay and scoliosis, requiring non-invasive ventilation while ambulant [14]. Muscle biopsies showed fibre type disproportion. However, the molecular mechanisms underlying the muscle fibre dysfunction caused by this mutation are unknown.
2. Methodical approach and research results
We studied the effect of the E173A mutation in recombinant Tpm3.12 on actin-myosin interaction at different simulated stages of the ATPase cycle (±Ca2+). Conformational changes in actin, myosin head (S1) and tropomyosin modified by fluorescent probes were studied in the ghost muscle fibres using polarized fluorimetry, a well-established technique for this application [15][16].
The results show that tropomyosin with E173A replacement affects the proportion of the switched-on and switched-off actin monomers, the balance of myosin subfragment-1 (S1) strongly and weakly bound with F-actin, and the tropomyosin position during the ATPase cycle. It is assumed that the E173A mutation weakens the ability of troponin-C to switch actin monomers on and activates the strong binding of the myosin heads to F-actin at low Ca2+ by suppressing the troponin-I ability to switch actin monomers off, and also induces the appearance of the strongly bound myosin heads (the rigor-like myosin heads) at relaxation, which may be one of the causes of muscle weakness. We suggest that replacing negatively charged glutamate 173 with neutral hydrophobic alanine may cause the salt bridge between tropomyosin residues 173 and 169 to break, and as a result, partially destabilize the tropomyosin molecule in the region of the site for tropomyosin binding to troponin-T. The alteration in the tropomyosin to troponin-T interaction can result in disruption of the ability of troponin to switch the thin filaments on and off. This can lead to inhibition of the ATPase activity at high Ca2+ (a decrease in force production) and increase in the Ca2+-sensitivity and the appearance of the rigor-like myosin heads at relaxation. The so-called rigor-like cross-bridges can be one of the reasons for contractures and disorganization of muscle fibres.
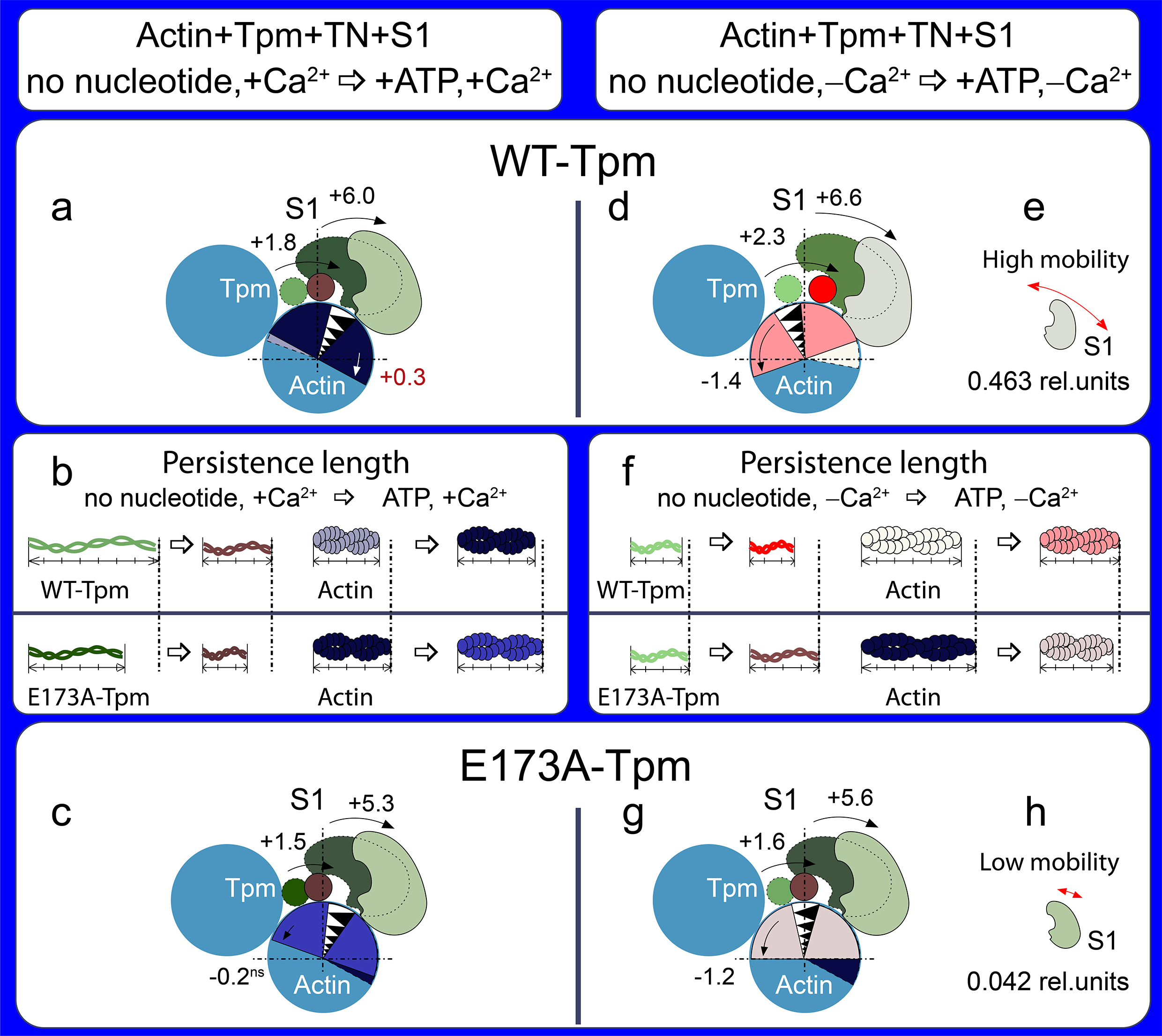
It seems important to reduce the effect of the E173A mutation. The Ca2+-desensitizer W7 is able to restore the ability of troponin to activate the strong binding of the myosin heads to F-actin at high Ca2+ and to reduce the number of rigor-like myosin heads at relaxation. However, W7 does not restore the ability of troponin to switch the thin filaments off at low Ca2+. Therefore, W7 can be used more likely to reduce the damaging effect of the E173A mutation on muscle contractility.
References
- A. M. Gordon; E. Homsher; Michael Regnier; Regulation of contraction in striated muscle.. Physiological Reviews 2000, 80, 853-924, 10.1152/physrev.2000.80.2.853.
- R. Craig; W. Lehman; Crossbridge and tropomyosin positions observed in native, interacting thick and thin filaments11Edited by W. Baumeister. Journal of Molecular Biology 2001, 311, 1027-1036, 10.1006/jmbi.2001.4897.
- R. Craig; W. Lehman; Crossbridge and tropomyosin positions observed in native, interacting thick and thin filaments11Edited by W. Baumeister. Journal of Molecular Biology 2001, 311, 1027-1036, 10.1006/jmbi.2001.4897.
- X. Li; W. Lehman; S. Fischer; The relationship between curvature, flexibility and persistence length in the tropomyosin coiled-coil. Journal of Structural Biology 2010, 170, 313-318, 10.1016/j.jsb.2010.01.016.
- W. Lehman; X. Li; F.A. Kiani; J.R. Moore; S.G. Campbell; S. Fischer; M.J. Rynkiewicz; Precise Binding of Tropomyosin on Actin Involves Sequence-Dependent Variance in Coiled-Coil Twisting. Biophysical Journal 2018, 115, 1082-1092, 10.1016/j.bpj.2018.08.017.
- Y.S. Borovikov; O.E. Karpicheva; S.V. Avrova; C.S. Redwood; Modulation of the effects of tropomyosin on actin and myosin conformational changes by troponin and Ca2+. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2009, 1794, 985-994, 10.1016/j.bbapap.2008.11.014.
- Y.S. Borovikov; N.A. Rysev; O.E. Karpicheva; V.V. Sirenko; S.V. Avrova; A. Piers; C.S. Redwood; Molecular mechanisms of dysfunction of muscle fibres associated with Glu139 deletion in TPM2 gene. Scientific Reports 2017, 7, 16797, 10.1038/s41598-017-17076-9.
- Y.S. Borovikov; O.E. Karpicheva; A.O. Simonyan; S.V. Avrova; E.A. Rogozovets; V.V. Sirenko; C.S. Redwood; The Primary Causes of Muscle Dysfunction Associated with the Point Mutations in Tpm3.12; Conformational Analysis of Mutant Proteins as a Tool for Classification of Myopathies. International Journal of Molecular Sciences 2018, 19, 3975, 10.3390/ijms19123975.
- Y. Yamada; K. Namba; T. Fujii; Cardiac muscle thin filament structures reveal calcium regulatory mechanism. Nature Communications 2020, 11, 1-9, 10.1038/s41467-019-14008-1.
- E. Behrmann; M. Müller; P.A. Penczek; H.G.Mannherz; D.J. Manstein; S. Raunser; Structure of the Rigor Actin-Tropomyosin-Myosin Complex. Cell 2012, 150, 327-338, 10.1016/j.cell.2012.05.037.
- S.V. Perry; Vertebrate tropomyosin: distribution, properties and function.. Journal of Muscle Research and Cell Motility 2001, 22, 5-49, 10.1023/a:1010303732441.
- M. Marttila; V-L. Lehtokari; S. Marston; T.A. Nyman; C. Barnerias; A.H. Beggs; E. Bertini; O. Ceyhan-Birsoy; P. Cintas; M. Gérard; et al.B. Gilbert-DussardierJ.S. HogueC. LongmanB. EymardM. FrydmanP.B. KangL. KlingeH. KolskiH. LochmüllerL. MagyV. ManelM. MayerE. MercuriK. N. NorthS. Peudenier-RobertH. PihkoF.J. ProbstR. ReisinW. StewartA.L. TaratutoM. VisserE. WilichowskiJ. WinerK. NowakN.G. LaingT.L. WinderN. MonnierN.F. ClarkeK. PelinM. GrӧnholmC. Wallgren-PetterssonO. Ceyhan-Birsoy Mutation update and genotype-phenotype correlations of novel and previously described mutations in TPM2 and TPM3 causing congenital myopathies.. Human Mutation 2014, 35, 779-90, 10.1002/humu.22554.
- C.A. Sewry; C. Wallgren‐Pettersson; Myopathology in Congenital Myopathies. Neuropathology and Applied Neurobiology 2017, 43, 5-23, 10.1111/nan.12369.
- P. Munot; D. Lashley; Heinz Jungbluth; L. Feng; M. Pitt; Stephanie A. Robb; J. Palace; S. Jayawant; R. Kennet; D. Beeson; et al.T. CullupS. AbbsNigel G. LaingC. SewryFrancesco Muntoni Congenital fibre type disproportion associated with mutations in the tropomyosin 3 (TPM3) gene mimicking congenital myasthenia. Neuromuscular Disorders 2010, 20, 796-800, 10.1016/j.nmd.2010.07.274.
- J. Borejdo; Priya Muthu; John Talent; Irina Akopova; Thomas P. Burghardt; Rotation of actin monomers during isometric contraction of skeletal muscle. Journal of Biomedical Optics 2007, 12, 014013-014013-10, 10.1117/1.2697286.
- Y.S. Borovikov; Conformational changes of contractile proteins and their role in muscle contraction.. Natural and Engineered Resistance to Plant Viruses, Part II 1999, 189, 267-301, 10.1016/s0074-7696(08)61389-3.

