 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Abolfazl Zarjou | + 4535 word(s) | 4535 | 2021-08-03 05:51:33 | | | |
| 2 | Amina Yu | Meta information modification | 4535 | 2021-08-11 03:29:40 | | |
Video Upload Options
As it pertains to the kidney, several clinical conditions have been recognized that are associated with significant amount of free heme and subsequent kidney damage. The kidney is frequently involved during clinical settings, with the common denominator of increased heme burden given its primary function of filtration. Moreover, the proximal tubules possess a high number of mitochondria that upon injury release their cytochrome heme content leading to higher levels of local heme and hence potentiating the cycle of injury.
1. Introduction
Heme is an evolutionarily conserved, ubiquitous iron-containing compound that is essential in numerous cellular functions [1][2][3]. However, heme is strongly hydrophobic and hence can readily enter cell membranes increasing cellular susceptibility to oxidant-mediated damage based on its reactivity and pro-oxidant properties [2][4]. There are hundreds of heme proteins, and heme is present in virtually all cellular compartments as a requisite for aerobic life [1]. The vast majority of heme in mammalians is confined within hemoglobin and myoglobin. Under homeostatic conditions cellular heme levels are stringently controlled, a process that involves a well-orchestrated balance between heme biosynthesis and catabolism. However, during pathological conditions and upon injury, heme can be released and when present in sufficient amount leads to commencement of an injury cycle that could eventually lead to cellular death and organ failure [2]. As it pertains to the kidney, several clinical conditions have been recognized that are associated with significant amount of free heme and subsequent kidney damage [5][6][7]. The kidney is frequently involved during clinical settings, with the common denominator of increased heme burden given its primary function of filtration. Moreover, the proximal tubules possess a high number of mitochondria that upon injury release their cytochrome heme content leading to higher levels of local heme and hence potentiating the cycle of injury. Although the list of such clinical settings is vast and includes some frequent diseases and syndromes, massive intravascular hemolysis and rhabdomyolysis are the main culprits that may result in diverse forms of kidney disease, most commonly acute kidney injury (AKI) [5]. In fact, both myoglobin and hemoglobin are filtered by the glomerulus into the urinary space where they are degraded, thus releasing heme pigment. As a defense against such toxicity cells promptly upregulate heme oxygenase-1 (HO-1) and ferritin expression [5][8]. HO catalyzes the regiospecific and rate limiting step of heme degradation to carbon monoxide (CO), ferrous iron and biliverdin [9]. Biliverdin is subsequently converted to bilirubin by the action of biliverdin reductase, while ferrous iron stimulates the induction of ferritin. In mammalian systems, two distinct enzymes make up the HO family; namely HO-1 and HO-2, and these enzymes are products of distinct genes [7]. Furthermore, while HO-1 is rapidly induced in response to a number of stimuli, HO-2 expression is constitutive. Discoveries in the early 1990s that described anti-oxidant and protective attributes of HO-1 [10] and ferritin [11] has led to a remarkable and vast field of investigations with many exciting and seminal breakthroughs. The cytoprotective effects of HO-1 induction are primarily due to exerting anti-inflammatory, anti-apoptotic, anti-oxidant, and anti-proliferative effects as well as modulation of fibrosis and neovascularization [3][4][5][12]. Within the limitation of the review, we will only discuss some of the most common conditions associated with increased heme burden from a clinical perspective and will follow that with brief description of the literature about current understanding of the molecular mechanisms that confer protection against heme mediated toxicity and organ damage.
2. Clinical Significance
Bywaters and Beall documented the first description of the consequences of traumatic muscle injury on kidney function during air-raid casualties of World War II [13]. They documented four patients who suffered crush injuries with similar clinical features that included brown-black granular casts, oliguria, progressive rise in blood urea and potassium and eventual death. Bywaters and Stead were also able to reproduce these findings by injection of human myoglobin in rabbits on an “acidifying” diet with urinary pH of below 6.0 [14]. In simple terms, rhabdomyolysis is the necrosis of muscle and subsequent release of intracellular components into circulation. It is now evident that rhabdomyolysis and resultant myoglobinuric AKI is a common and potentially life-threatening syndrome characterized by release of toxic muscle cell contents into the circulation [15][16][17][18]. On the whole, the degree of muscle damage rather than initiating factor may determine the clinical course of rhabdomyolysis ranging from asymptomatic illness to a life-threatening condition. While the etiological range of rhabdomyolysis is broad and pathological sequelae is complex (summarized in Figure 1 ), the presence of multiple concomitant etiologic factors is not uncommon [19].
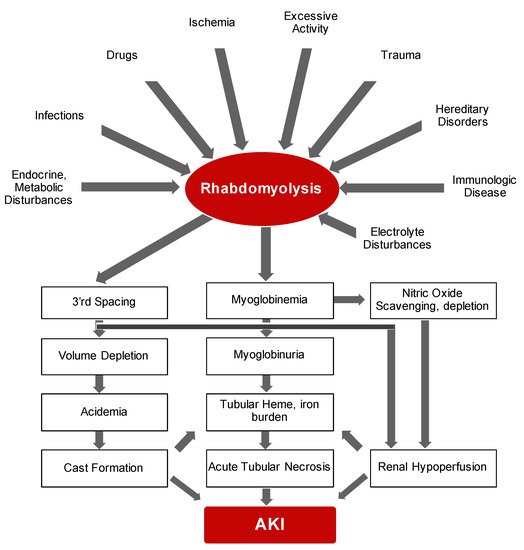
The major instigating factor of AKI is the release of myoglobin. The half-life of myoglobin is rather short mainly because it is not significantly protein-bound and hence is rapidly cleared by the kidneys and generally vanishes within six hours of release [20]. Therefore, the preferred diagnostic method of rhabdomyolysis is detection of creatine kinase, an intracellular enzyme, which has a serum half-life of around 36 h [21]. In addition to AKI, fluid and electrolyte abnormalities may also be noted during early presentation. With significant degree of muscle injury, extracellular fluid may shift into the damaged cells resulting in potential hypovolemia and hence further susceptibility to AKI. Release of high levels of intracellular phosphorus and potassium elevates their levels in circulation while dystrophic calcification may also lead to hypocalcemia. In addition, significant release of purines from damaged muscle cells and reduced kidney function, leading to diminished excretion, may also result in hyperuricemia. Disseminated intravascular coagulation, albeit rare, may be another consequence of severe rhabdomyolysis that is suggested to result from release pro-thrombotic molecules from damaged cells [22]. Taken together, rhabdomyolysis is a frequent, and—from an etiologic standpoint—a multifaceted clinical syndrome that requires prompt recognition and diagnosis and management of fluid and electrolyte abnormalities.
Shortened survival of red blood cells (RBCs) may be the result of either inherent abnormalities in RBCs (intracorpuscular), which frequently result from genetic defects, or external causes (extracorpuscular) that are generally acquired (partial list of causes of hemolytic anemias is summarized in Table 1 ).
| Causes of Hemolytic Anemias |
|---|
| Intrinsic (intracorpuscular) A. Genetic defects
Extrinsic (extracorpuscular) A. Antibody mediated
D. Chemical agents: Arsine, Glycerol, Benzene, Sodium chlorate, Methyl chloride, etc. E. Venoms: Rattle snake, Coral snake, Brown recluse spider |
PNH is a rare, potentially life-threatening hematopoietic stem cell condition leading to a myriad of clinical manifestations [23][24][25][26]. Given the low incidence and mild, nonspecific initial presentation, accurate diagnosis and management is delayed in many cases. The vast majority of cases are due to reduction in or absence of glycosylphosphatidylinositol-anchor on the cell surface [23][26]. This anchor plays a crucial role in linking multiple proteins to the plasma membrane of hematopoietic cells including blood group antigens and complement mediator proteins. Deficiency of these proteins renders the RBCs vulnerable to excessive complement damage and hemolysis. In particular, loss of CD55 and CD59 complement regulators is recognized as the main cause leading to several manifestations of the disease that includes intravascular hemolysis and kidney damage [26][27]. Lack of CD55 is followed by increased opsonization of RBCs and subsequent phagocytosis and removal by reticuloendothelial cells in an extravascular manner. In contrast, the absence of CD59 leads to unrestrained formation of the membrane attack complex of complement system followed by a classic type II hypersensitivity damage to RBCs and intravascular hemolysis [27]. Importantly, kidney disease has been proposed to be a leading cause of mortality in this group of patients. Kidney disease associated with PNH was first described in 1971 [28]. A subsequent study of 21 patients with PNH revealed substantial incidence of functional and anatomic renal abnormalities encompassing hyposthenuria, tubular dysfunction, and declining glomerular filtration rate [29]. Importantly, pathological findings are very similar to sickle cell disease, which underscores the increased heme and hemosiderosis burden in the kidneys. This notion is also supported by presence of hemolysis in almost all patients that presented with AKI and PNH [30]. While these facts highlight the underlying unfettered heme as the main culprit of kidney disease in patients with PNH, it is obvious that the nonspecific and variable presentation and degree of hemolysis account for an overall lack of clinical reports of renal involvement in PNH [31]. As mentioned, clinical presentation of PNH is highly variable and dependent on multiple factors, and hence requires a high degree of suspicion for accurate diagnosis [32][33]. The presenting symptoms may include fatigue, chest pain, hemoglobinuria, dyspnea, abdominal pain, thrombosis and renal dysfunction, among others [33]. Once PNH is suspected, flow cytometry has emerged as the main diagnostic tool where patient’s peripheral blood cells are analyzed for reduction in or absence of CD55 and CD59, among other GPI-anchored proteins [34]. Treatment of PNH is based on the specific PNH category which itself is primed by the severity of hemolysis-associated symptoms and degree of bone marrow failure.
3. Molecular Insights
Heme is a complex of iron with protoporphyrin IX with fundamental biological functions for all aerobic cells. The inimitable function of heme is evidenced by its synthesis in all nucleated cells. By serving as the prosthetic group of many hemoproteins, it participates in diverse biological functions ranging from electron transportation, gas carriage, gas sensors and working as a cellular messenger [7][35][36]. Apart from hemoglobin and myoglobin, many other proteins also depend on heme to carry their vital functions that include cytochrome P450 enzymes, cytochromes involved in mitochondrial respiration, nitric oxide synthasesas, catalases, and peroxidases, histidine kinases, nitrophorins, among others [2][35]. Biosynthesis of heme is complex, involves multiple enzymes that work in concert inside the mitochondria and cytoplasm with the mitochondrial δ-aminolevulinic acid synthase serving as the rate-limiting enzyme [7]. The biosynthesis of heme is also heavily dependent on the rate of heme catabolism. Under physiological conditions amount of free heme is minimal and multiple evolutionary mechanisms have evolved to curtail availability of free heme following its release after injury. Given the vast amount of heme present in hemoglobin and myoglobin, it is not surprising that hemolysis and rhabdomyolysis are the two main culprits that lead to significant release of heme from damaged cells [37]. Notably, extracellular hemoglobin and myoglobin are easily oxidized leading to the transition of iron from a ferrous to a ferric state within the heme prosthetic group and subsequent release of heme culminating in significant rise of free heme [38][39][40]. Oxidation of heme moieties in hemoglobin to ferryl states also occur in vivo [41]. Ferric hemoglobin was demonstrated to lose heme moieties more readily than ferryl hemoglobin [42]. The extracellular hemoglobin and heme are readily scavenged by haptoglobin and hemopexin, respectively [43]. Ferryl hemoglobin is taken up via phagocytosis as well as CD163 receptor-mediated endocytosis [41]. The significance of scavenging free heme and the subsequent mitigation of heme’s injurious effects is well documented in a number of clinical settings, including, but not limited to, sickle cell disease and malaria [44][45][46][47]. These studies provide irrefutable evidence that not only support the injurious effects of free heme but also strategies that improve clinical outcomes by scavenging unfettered heme. However, this scavenging capacity can become exhausted, and subsequently free heme can accumulate in circulation. The hydrophobic nature of heme allows for its passage across cell membranes and subsequent buildup within the hydrophobic milieu of intact cells. A strong body of evidence demonstrates that free heme can cause direct and indirect injurious effects [4][48][37][40][49]. Such injurious effects are mediated by the pro-oxidant, pro-inflammatory and cytotoxic properties of heme. The pro-oxidant attributes of heme are multifaceted and include the generation of alkoxyl and peroxyl radicals that can further trigger lipid peroxidation [50][51]. Additionally, iron at the core of heme can participate in the Fenton reaction with subsequent formation of hyroxy-radicals [52]. Furthermore, heme also contributes to the formation of free radicals via enzymatic reactions of NADPH oxidases [4]. The peroxides generated during the mitochondrial respiration may also aggravate the pro-oxidant effects of heme [53]. The pro-inflammatory effects of heme are also well recognized [54][55][56][57]. While macrophages have been shown to secrete tumor necrosis factor in response to addition of heme in vitro [58], in vivo administration of heme results in increased vascular permeability, adhesion molecule expression, and leukocyte recruitment [57]. Furthermore, heme can also induce monocyte chemoattractant protein-1 [56] and enhance endothelial cell adhesion molecule expression [59], causing the activation of polymorphonuclear leukocytes and endothelial uptake of heme results in heightened damage by these activated cells [60][61]. The interplay between heme and macrophage-mediated inflammatory response and consequent kidney injury was elegantly described in a model of rhabdomyolysis in mice. This study demonstrated how macrophages contribute to the pathogenesis of rhabdomyolysis by releasing extracellular traps that are comprised of DNA fibers and granule proteins. Importantly, heme-activated platelets released from necrotic muscle cells during rhabdomyolysis augmented the production of these extracellular traps through increasing intracellular ROS formation as well as histone citrullination [62]. The relationship between heme and complement activation during rhabdomyolysis is also documented. Specifically, evidence supports a key role for myoglobin-derived heme activation of the alternative pathway arm of complement system [63]. It is evident that heme is a potent damage-associated molecular pattern. Exposure of neutrophils to heme is associated with multiple inflammatory characteristics. These include the organization of cytoskeleton in a manner that is consistent with phagocytic and migratory activities, increase in generation of neutrophil extracellular trap, generation of ROS via NADPH oxidase activity, upregulation of neutrophil survival factors such as IL-8, and stimulation of ERK, PI3K–Akt and nuclear factor-κB signaling pathways, all of which indisputably prolong inflammation [49][64][65]. Overall, while heme is a cardinal necessity for aerobic life, a strong body of evidence points towards its potential hazardous effects and obviates the demand for an overhaul mechanism to mitigate these effects, namely the HO-1/ferritin system.
Heme oxygenases (HO) are comprised of two isozymes, HO-1 and HO-2, that are responsible for catalyzing degradation of heme into ferrous iron, CO and biliverdin, the latter subsequently converted to bilirubin by the enzymatic reaction of biliverdin reductase [12]. While the expression of HO-1 is induced in response to a variety of endogenous and exogenous stimuli, HO-2 is a constitutive isoform of HO family and is expressed largely in the brain, kidney, and testis [12]. The byproducts of HO reaction were initially considered to be mere waste products. However, interest and robust investigations in the past three decades have affirmed that CO, biliverdin, bilirubin and iron induced ferritin expression are important mediators of many biological functions such as apoptosis, autophagy, immune cell trafficking, mitochondrial homeostatic integrity, regulation of innate and adaptive immunity, regulation of cell cycle, and angiogenesis, among others [5][6][12]. These observations were made possible thanks to a number of seminal breakthroughs. First, Nath and colleagues reported that the induction of HO-1 coupled to ferritin synthesis is a rapid, protective antioxidant response in a model of rhabdomyolysis induced AKI [10]. This was followed by observations reported by Balla and colleagues that induction of ferritin was protective in endothelial cells that were exposed to oxidant mediated injury [11]. Next, the generation of HO-1 knockout mice that evinced multiple pathological manifestations further substantiated the relevance of this enzyme system in the context of health and disease and delivered a platform for the investigations that followed [66]. The first description and diagnosis of a young patient with HO-1 deficiency that was reported by Yachie and colleagues revealed many similarities to the HO-1 knockout mice [67]. This rare, autosomal recessive disease has been since reported in a small number of other cases that underscores the indispensable nature of this enzyme [68]. The protective effects of HO-1 induction in diseases leading to increased heme burden are intricate. Importantly, under physiological conditions, the expression of HO-1 is suppressed via transcription factor, Bach-1 [69]. Induction of HO-1 expression not only leads to prompt degradation and removal of pro-oxidant and pro-inflammatory heme, but also generates molecules that are now well recognized to possess cytoprotective properties. Notably, there is also evidence that these cytoprotective effects may also be independent from the enzymatic activity of HO-1 and may also rely on nuclear localization and activation of transcription factors essential during oxidative stress [70]. As it relates to the immune system, it was demonstrated that macrophages lacking HO-1 expression are able to perform erythrophagocytosis, but this was followed by the rupture and death of macrophages leading to the release of oxidized heme and worsening inflammation [71]. The relevance of myeloid HO-1 expression is highlighted in other studies where it was shown that myeloid HO-1 controls the activation of interferon-regulatory factor-3 after Toll-like receptor 3 or 4 stimulation, or viral infection, suggesting that HO-1 plays a critical function in innate immunity [72]. The generated CO exerts beneficial effects in a number of injury models including hyperoxia, ischemia-reperfusion mediated injury, graft rejection, malaria, and sepsis among others [73][74]. Many cellular mechanisms have been proposed that mediated such favorable outcomes. CO is known to exert anti-inflammatory, anti-apoptotic and pro-phagocytic properties [74]. CO is shown to induce autophagy [75], inhibit mitochondrial dysfunction and inflammasome activation in macrophages [76], modulate mitochondrial biogenesis [77], and enhance acceleration of resolution of inflammation through biosynthesis of specialized pro-resolving mediators [78]. Biliverdin, another byproduct of the HO enzymatic reaction, is readily converted to bilirubin by the action of biliverdin reductase. Several salutary effects, anti-mutagenic, an antioxidant, anti-inflammatory, and immunosuppressant have been attributed to biliverdin [79]. Additionally, bilirubin has long been recognized for its potent anti-oxidant properties [80]. Overall, the cytoprotective properties of biliverdin and bilirubin have been established in a number of injury settings that include ischemia-reperfusion, graft rejection, sepsis, and intimal hyperplasia [81][82]. Ferritin also plays a paramount protective role in injurious settings. The ferritins are a family of proteins characterized by highly conserved three-dimensional structures similar to spherical shells, designed to sequester and store large amounts of iron in a safe, soluble and bioavailable form. It is made of 24 subunits of two types (FtH, heavy and FtL, light chain) whose proportion depends on the iron status of the cell, the tissue and the organ [8][83]. The indispensable functions of FtH are underscored by the embryonic lethality of mice with global deletion of FtH [84]. We and others have demonstrated the unique nephroprotective role of H-ferritin in different models of injury as well as its role in mitigating osteoblastic transition of cells that are evoked by uremic milieu [85][86][87][88]. Moreover, it must be noted that global HO-1 deletion, and targeted deletion of HO-1 and FtH in proximal tubules did not result in any significant abnormality in kidney function, as evidenced by normal serum creatinine levels as well as lack of proteinuria. Nevertheless, deletion of HO-1 and FtH is accompanied by substantial susceptibility to kidney disease in different forms of injury models [88][89][90][91].
4. Evidence to Support Salutary Effects of HO-1/Ferritin System against Heme-Protein Induced Kidney Disease
The first line of defense against potential toxicity of unstable, oxidized hemoglobin and heme following hemolysis are haptoglobin and hemopexin, respectively [43]. However, as discussed earlier, this line of defense can be overwhelmed and saturated during massive hemolysis leading to increasing levels of free heme. It is of note that, to the best of our knowledge, no protein has been identified with the capacity to bind myoglobin following its release from disrupted muscle. Evidence that supports the crucial role of HO-1 has accumulated over the past three decades. Using hypertonic glycerol to induce rhabdomyolysis, Nath and colleagues were the first to demonstrate that inhibition of HO activity resulted in heightened injury, while prior conditioning of the animals with hemoglobin conferred protection [10]. These observations were later supported by similar experiments in animals with global deletion of HO-1 where deficiency of HO-1 was coupled with irreversible AKI and 100% mortality [92]. Importantly, an eight-fold increment in renal content of heme was observed in knockout animals, which further corroborates the adverse effects of free heme. In another study, using humanized HO-1 mice, where the human HO-1 gene, along with all its regulatory region, was inserted into mice with global HO-1 deficiency, it was revealed that the human HO-1 was functional, able to rescue the phenotype of HO-1 deficiency and conferred significant protection against rhabdomyolysis-induced AKI [93]. Wei and colleagues sought to investigate the mechanisms that govern the beneficial effects of granulocyte colony-stimulating factor (G-CSF) and found that such effects of G-CSF during glycerol induced AKI were dependent on HO-1. They found that G-CSF potently induced HO-1 in both cultured tubular cells and mouse kidneys, and that chemical inhibition of HO-1 was associated with mitigation of the protective effects of G-CSF [94]. Boddu et al. described a leucine-rich repeat kinase 2 (Lrrk2) deletion model of Parkinson’s disease in rats, wherein hemoglobin accumulation in kidney results in concomitant induction of HO-1. The Lrrk2 knockout rat model was protected against glycerol-induced rhabdomyolysis and preconditioning of these rats with endogenous hemoglobin conferred protection against AKI [95]. The protective effects of curcumin (active component in turmeric rhizomes and a known anti-oxidant), was also shown to at least in part confer protection during glycerol induced AKI via the upregulation of HO-1 [96]. The relevance of ferritin expression in the context of protective effects of HO-1 induction was highlighted in a study with targeted deletion of H-ferritin in proximal tubules (more details below) [88]. The non-redundant protective role of ferritin and hazardous role of free iron are supported by studies using deferoxamine (DFO), a potent iron cations chelator. Shah and colleagues demonstrated the negative effects of hydroxyl radical in glycerol-induced AKI and established that limiting iron availability via DFO was protective against AKI in this model [97]. Similarly, Paller’s studies showed that DFO administration was protective in two experimental models of pigment-induced AKI, intramuscular glycerol injection and intravenous hemoglobin infusion without and with concurrent ischemia in the rat [98].
HO-1 induction and its corresponding advantageous effects were first shown to protect against experimental cerebral malaria [99]. In this study, genetic deletion of HO-1 and its enzymatic inhibition were associated with a higher incidence of cerebral malaria. Conversely, pharmacological induction of HO-1 and CO administration reduced such incidence and diminished blood–brain barrier disruption, brain microvasculature congestion and neuroinflammation. Mechanistic studies revealed that the binding capability of CO to cell-free Hb preventing heme release is a major underlying mechanism of these protective effects [99]. These findings and other investigations strongly indicate that free heme is a major culprit of severe malaria [47]. These protective effects have been validated in other studies and extend to mitigation of acute lung injury [100] as well as clinical outcome in pregnancy-associated malaria [101]. More recently, using various transgenic models, it was demonstrated that the development of disease tolerance to malaria is achieved via a damage control mechanism operating specifically in proximal tubular cells. Such tolerance was shown to be dependent on HO-1 and FtH expression, a mechanism that implicates the transcription-factor nuclear-factor E2-related factor-2 (NRF2). Overall, proximal tubular upregulation of HO-1 and H-ferritin detoxify free heme in proximal tubules thus alleviating development of a hallmark of severe malaria, namely AKI [102].
Vaso-occlusion and hemolysis are the clinical hallmarks of SCD and patients with SCD are at increased risk of developing chronic kidney disease [103]. As a consequence of frequent hemolysis, triggered by sickling (formation of rigid strands within the RBC that gives its characteristic, sickle shape) of affected RBCs, free hemoglobin and heme are released into circulation. During the initial phase, haptoglobin and hemopexin confer the first line of protection, but once saturated, the levels of free hemoglobin and heme significantly rise in the blood of SCD patients [104][105]. Nath and colleagues established that transgenic sickle mice display renal enlargement, increased heme content and HO activity, as well as medullary congestion. The human sickle kidney also shows similar findings with induction of HO-1 in renal tubules, interstitial cells, and in the vasculature [106]. Other investigators have validated these results by demonstrating HO-1 induction in different SCD mouse models [107][108]. Belcher and colleagues provided additional evidence to support the pivotal role of HO-1 in prevention of vaso-occlusion in transgenic sickle mice [109]. While there was a systematic induction of HO-1, treatment with hemin resulted in further overexpression of HO-1 accompanied by a reduction in hemostasis, leukocyte-endothelium interactions, and inhibition of some key pro-inflammatory proteins such as NF-κB, VCAM-1, and ICAM-1 expression [109]. Furthermore, while the pharmacological inhibition of HO activity led to the worsening stasis in these mice, protective effects of HO-1 induction were mimicked by HO byproducts biliverdin and CO. In fact, it has been postulated that increased levels of CO are the main driving force behind hyperperfusion and hyperfiltration which is frequently observed in SCD patients [110][111]. These effects are likely due to offsetting the vasoconstriction associated with nitric oxide depletion [112]. In humans, a variable GT-repeat length polymorphism in the HO-1 gene promoter is inversely correlated to HO-1 expression [113]. Saraf and colleagues demonstrated that in patients with SCD, homozygosity of the long allele (leading to lower levels of HO-1 expression) was associated with decreased glomerular filtration rate. In addition, the authors found enrichment of single nucleotide polymorphism downstream of HO-1 in sickle patients that was associated with chronic kidney disease stage or end stage kidney disease [105].
Increasing heme burden leads to the eventuality of increased iron release from heme moiety. Iron, the second most abundant element on earth, has the ability to change its valence and hence serve as a unique contributor in multiple diverse biological pathways. The ferrous iron released from heme degradation can readily participate in Fenton’s reaction, leading to the generation of ROS, which may induce the oxidation of proteins, lipids and lipoproteins, nucleic acids, carbohydrates and other cellular components [52]. The ability of free iron to exacerbate kidney injury is well documented as its chelation via deferoxamine is shown to be protective in injury models that are coupled with increased heme burden [97][98]. Moreover, Zager and colleagues delivered evidence using different models of kidney injury that upregulation of ferritin may serve as an advantageous “preconditioning” state [49][114]. There is also mounting clinical substantiation to suggest that higher plasma catalytic iron levels are associated with worse outcomes including AKI and need for kidney replacement therapy [115][116]. We have shown that targeted deletion of FtH in proximal tubules leads to heightened injury in glycerol-induced rhabdomyolysis, as supported by higher serum creatinine and mortality, as well as worse histological manifestations. Intriguingly, we also showed that deletion of FtH resulted in markedly higher levels of HO-1 expression, particularly post-injury [86]. Despite this increase in HO-1 expression, mice with deletion of proximal tubule-specific FtH succumbed significantly more frequently following rhabdomyolysis. This observation unequivocally demonstrates that the salutatory effects of HO-1 expression are co-dependent on FtH. These findings are corroborated in another study where FtH overexpression, through doxycycline-inducible system, led to lesser apoptosis and improved tubular viability, likely through mitigating the oxidative stress [117]. Moreover, it has been shown that systemic and locally generated hepcidin protect against hemoglobin-induced AKI. Through subsequent studies, Scindia and colleagues recapitulated the protective effects of hepcidin in AKI and postulated that hepcidin exerts such effects through increased iron retention with subsequent upregulation of intracellular ferritin [118]. Similar results have been recapitulated in a model of hemoglobin-mediated nephropathy [119]. As mentioned above, recent studies underscored the significance of proximal tubular HO-1 and FtH induction in a malaria model. Using transgenic mice with targeted deletion of FtH or HO-1, it was shown that these proteins are critical to establish disease tolerance to malaria by detoxifying the free heme and diminishing development of AKI [102].
References
- Paoli, M.; Marles-Wright, J.; Smith, A. Structure-function relationships in heme-proteins. DNA Cell Biol. 2002, 21, 271–280.
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354.
- Balla, J.; Vercellotti, G.M.; Nath, K.; Yachie, A.; Nagy, E.; Eaton, J.W.; Balla, G. Haem, haem oxygenase and ferritin in vascular endothelial cell injury. Nephrol. Dial. Transplant. 2003, 18 (Suppl. 5), v8–v12.
- Bozza, M.T.; Jeney, V. Pro-inflammatory actions of heme and other hemoglobin-derived DAMPs. Front. Immunol. 2020, 11, 1323.
- Bolisetty, S.; Zarjou, A.; Agarwal, A. Heme oxygenase 1 as a therapeutic target in acute kidney injury. Am. J. Kidney Dis. 2017, 69, 531–545.
- Zarjou, A.; Agarwal, A. Heme oxygenase-1 as a target for TGF-beta in kidney disease. Semin. Nephrol. 2012, 32, 277–286.
- Tracz, M.J.; Alam, J.; Nath, K.A. Physiology and pathophysiology of heme: Implications for kidney disease. J. Am. Soc. Nephrol. 2007, 18, 414–420.
- Balla, J.; Balla, G.; Zarjou, A. Ferritin in kidney and vascular related diseases: Novel roles for an old player. Pharmaceuticals 2019, 12, 96.
- Nath, K.A. Heme oxygenase-1 and acute kidney injury. Curr. Opin. Nephrol. Hypertens. 2014, 23, 17–24.
- Nath, K.A.; Balla, G.; Vercellotti, G.M.; Balla, J.; Jacob, H.S.; Levitt, M.D.; Rosenberg, M.E. Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J. Clin. Investig. 1992, 90, 267–270.
- Balla, G.; Jacob, H.S.; Balla, J.; Rosenberg, M.; Nath, K.; Apple, F.; Eaton, J.W.; Vercellotti, G.M. Ferritin: A cytoprotective antioxidant strategem of endothelium. J. Biol. Chem. 1992, 267, 18148–18153.
- Ayer, A.; Zarjou, A.; Agarwal, A.; Stocker, R. Heme Oxygenases in cardiovascular health and disease. Physiol. Rev. 2016, 96, 1449–1508.
- Bywaters, E.G.; Beall, D. Crush injuries with impairment of renal function. Br. Med. J. 1941, 1, 427–432.
- Bywaters, E.G.L.; Stead, J.K. The production of renal failure following injection of solutions containing myohæmoglobin. Q. J. Exp. Physiol. Cogn. Med Sci. 1944, 33, 53–70.
- Bosch, X.; Poch, E.; Grau, J.M. Rhabdomyolysis and acute kidney injury. N. Engl. J. Med. 2009, 361, 62–72.
- Aleckovic-Halilovic, M.; Pjanic, M.; Mesic, E.; Storrar, J.; Woywodt, A. From quail to earthquakes and human conflict: A historical perspective of rhabdomyolysis. Clin. Kidney J. 2021, 14, 1088–1096.
- Cote, D.R.; Fuentes, E.; Elsayes, A.H.; Ross, J.J.; Quraishi, S.A. A “crush” course on rhabdomyolysis: Risk stratification and clinical management update for the perioperative clinician. J. Anesth. 2020, 34, 585–598.
- Cabral, B.M.I.; Edding, S.N.; Portocarrero, J.P.; Lerma, E.V. Rhabdomyolysis. Dis. Mon. 2020, 66, 101015.
- Gabow, P.A.; Kaehny, W.D.; Kelleher, S.P. The spectrum of rhabdomyolysis. Medicine 1982, 61, 141–152.
- Garry, D.J.; Mammen, P.P. Molecular insights into the functional role of myoglobin. Adv. Exp. Med. Biol. 2007, 618, 181–193.
- Brancaccio, P.; Lippi, G.; Maffulli, N. Biochemical markers of muscular damage. Clin. Chem. Lab. Med. 2010, 48, 757–767.
- Huerta-Alardin, A.L.; Varon, J.; Marik, P.E. Bench-to-bedside review: Rhabdomyolysis—An overview for clinicians. Crit. Care 2005, 9, 158–169.
- Fattizzo, B.; Serpenti, F.; Giannotta, J.A.; Barcellini, W. Difficult cases of paroxysmal nocturnal hemoglobinuria: Diagnosis and therapeutic novelties. J. Clin. Med. 2021, 10, 948.
- Risitano, A.M. Paroxysmal nocturnal hemoglobinuria and the complement system: Recent insights and novel anticomplement strategies. Adv. Exp. Med. Biol. 2013, 735, 155–172.
- Savage, W.J.; Brodsky, R.A. New insights into paroxysmal nocturnal hemoglobinuria. Hematology 2007, 12, 371–376.
- Hill, A.; DeZern, A.E.; Kinoshita, T.; Brodsky, R.A. Paroxysmal nocturnal haemoglobinuria. Nat. Rev. Dis. Primers 2017, 3, 17028.
- Brodsky, R.A. Paroxysmal nocturnal hemoglobinuria. Blood 2014, 124, 2804–2811.
- Rubin, H. Paroxysmal nocturnal hemoglobinuria with renal failure. JAMA 1971, 215, 433–436.
- Clark, D.A.; Butler, S.A.; Braren, V.; Hartmann, R.C.; Jenkins, D.E., Jr. The kidneys in paroxysmal nocturnal hemoglobinuria. Blood 1981, 57, 83–89.
- Chow, K.M.; Lai, F.M.; Wang, A.Y.; Chan, Y.L.; Tang, N.L.; Li, P.K. Reversible renal failure in paroxysmal nocturnal hemoglobinuria. Am. J. Kidney Dis. 2001, 37, E17.
- Ram, R.; Adiraju, K.P.; Gudithi, S.; Dakshinamurty, K.V. Renal manifestations in paroxysmal nocturnal hemoglobinuria. Indian J. Nephrol. 2017, 27, 289–293.
- Parker, C.; Omine, M.; Richards, S.; Nishimura, J.; Bessler, M.; Ware, R.; Hillmen, P.; Luzzatto, L.; Young, N.; Kinoshita, T.; et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood 2005, 106, 3699–3709.
- Schrezenmeier, H.; Muus, P.; Socie, G.; Szer, J.; Urbano-Ispizua, A.; Maciejewski, J.P.; Brodsky, R.A.; Bessler, M.; Kanakura, Y.; Rosse, W.; et al. Baseline characteristics and disease burden in patients in the International Paroxysmal Nocturnal Hemoglobinuria Registry. Haematologica 2014, 99, 922–929.
- Borowitz, M.J.; Craig, F.E.; Digiuseppe, J.A.; Illingworth, A.J.; Rosse, W.; Sutherland, D.R.; Wittwer, C.T.; Richards, S.J.; Clinical Cytometry, S. Guidelines for the diagnosis and monitoring of paroxysmal nocturnal hemoglobinuria and related disorders by flow cytometry. Cytometry B Clin. Cytom. 2010, 78, 211–230.
- Ponka, P. Cell biology of heme. Am. J. Med. Sci. 1999, 318, 241–256.
- Fujiwara, T.; Harigae, H. Biology of heme in mammalian erythroid cells and related disorders. Biomed. Res. Int. 2015, 2015, 278536.
- Soares, M.P.; Bozza, M.T. Red alert: Labile heme is an alarmin. Curr. Opin. Immunol. 2016, 38, 94–100.
- Balla, J.; Vercellotti, G.M.; Jeney, V.; Yachie, A.; Varga, Z.; Jacob, H.S.; Eaton, J.W.; Balla, G. Heme, heme oxygenase, and ferritin: How the vascular endothelium survives (and dies) in an iron-rich environment. Antioxid. Redox Signal. 2007, 9, 2119–2137.
- Nagy, E.; Eaton, J.W.; Jeney, V.; Soares, M.P.; Varga, Z.; Galajda, Z.; Szentmiklosi, J.; Mehes, G.; Csonka, T.; Smith, A.; et al. Red cells, hemoglobin, heme, iron, and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1347–1353.
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercellotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879–887.
- Balla, J.; Potor, L.; Hendrik, Z.; Patsalos, A.; Katona, E.; Mehes, G.; Poliska, S.; Csosz, E.; Kallo, G.; Komaromi, I.; et al. Oxidation of hemoglobin drives a pro-atherogenic polarization of macrophages in human atherosclerosis. Antioxid. Redox Signal. 2021.
- Kassa, T.; Jana, S.; Meng, F.; Alayash, A.I. Differential heme release from various hemoglobin redox states and the upregulation of cellular heme oxygenase-1. FEBS Open Bio 2016, 6, 876–884.
- Smith, A.; McCulloh, R.J. Hemopexin and haptoglobin: Allies against heme toxicity from hemoglobin not contenders. Front. Physiol. 2015, 6, 187.
- Vinchi, F.; Costa da Silva, M.; Ingoglia, G.; Petrillo, S.; Brinkman, N.; Zuercher, A.; Cerwenka, A.; Tolosano, E.; Muckenthaler, M.U. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood 2016, 127, 473–486.
- Ashouri, R.; Fangman, M.; Burris, A.; Ezenwa, M.O.; Wilkie, D.J.; Dore, S. Critical role of hemopexin mediated cytoprotection in the pathophysiology of sickle cell disease. Int. J. Mol. Sci. 2021, 22, 6408.
- Vinchi, F.; De Franceschi, L.; Ghigo, A.; Townes, T.; Cimino, J.; Silengo, L.; Hirsch, E.; Altruda, F.; Tolosano, E. Hemopexin therapy improves cardiovascular function by preventing heme-induced endothelial toxicity in mouse models of hemolytic diseases. Circulation 2013, 127, 1317–1329.
- Ferreira, A.; Balla, J.; Jeney, V.; Balla, G.; Soares, M.P. A central role for free heme in the pathogenesis of severe malaria: The missing link? J. Mol. Med. 2008, 86, 1097–1111.
- Van Avondt, K.; Nur, E.; Zeerleder, S. Mechanisms of haemolysis-induced kidney injury. Nat. Rev. Nephrol. 2019, 15, 671–692.
- Haines, D.D.; Tosaki, A. Heme degradation in pathophysiology of and countermeasures to inflammation-associated disease. Int. J. Mol. Sci. 2020, 21, 9698.
- Vincent, S.H. Oxidative effects of heme and porphyrins on proteins and lipids. Semin. Hematol. 1989, 26, 105–113.
- Tappel, A.L. Unsaturated lipide oxidation catalyzed by hematin compounds. J. Biol. Chem. 1955, 217, 721–733.
- Zarjou, A.; Balla, J.; Balla, G.; Agarwal, A. Iron metabolism and oxidative stress. In Studies on Renal Disorders; Miyata, T., Eckardt, K.-U., Nangaku, M., Eds.; Humana Press: Totowa, NJ, USA, 2011; pp. 205–228.
- Nath, K.A.; Grande, J.P.; Croatt, A.J.; Likely, S.; Hebbel, R.P.; Enright, H. Intracellular targets in heme protein-induced renal injury. Kidney Int. 1998, 53, 100–111.
- Dutra, F.F.; Alves, L.S.; Rodrigues, D.; Fernandez, P.L.; de Oliveira, R.B.; Golenbock, D.T.; Zamboni, D.S.; Bozza, M.T. Hemolysis-induced lethality involves inflammasome activation by heme. Proc. Natl. Acad. Sci. USA 2014, 111, E4110–E4118.
- Ghosh, S.; Adisa, O.A.; Chappa, P.; Tan, F.; Jackson, K.A.; Archer, D.R.; Ofori-Acquah, S.F. Extracellular hemin crisis triggers acute chest syndrome in sickle mice. J. Clin. Investig. 2013, 123, 4809–4820.
- Nath, K.A.; Vercellotti, G.M.; Grande, J.P.; Miyoshi, H.; Paya, C.V.; Manivel, J.C.; Haggard, J.J.; Croatt, A.J.; Payne, W.D.; Alam, J. Heme protein-induced chronic renal inflammation: Suppressive effect of induced heme oxygenase-1. Kidney Int. 2001, 59, 106–117.
- Wagener, F.A.; Eggert, A.; Boerman, O.C.; Oyen, W.J.; Verhofstad, A.; Abraham, N.G.; Adema, G.; van Kooyk, Y.; de Witte, T.; Figdor, C.G. Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood 2001, 98, 1802–1811.
- Figueiredo, R.T.; Fernandez, P.L.; Mourao-Sa, D.S.; Porto, B.N.; Dutra, F.F.; Alves, L.S.; Oliveira, M.F.; Oliveira, P.L.; Graca-Souza, A.V.; Bozza, M.T. Characterization of heme as activator of Toll-like receptor 4. J. Biol. Chem. 2007, 282, 20221–20229.
- Wagener, F.A.; Feldman, E.; de Witte, T.; Abraham, N.G. Heme induces the expression of adhesion molecules ICAM-1, VCAM-1, and E selectin in vascular endothelial cells. Proc. Soc. Exp. Biol. Med. 1997, 216, 456–463.
- Balla, G.; Vercellotti, G.; Eaton, J.W.; Jacob, H.S. Heme uptake by endothelium synergizes polymorphonuclear granulocyte-mediated damage. Trans. Assoc. Am. Phys. 1990, 103, 174–179.
- Balla, G.; Vercellotti, G.M.; Muller-Eberhard, U.; Eaton, J.; Jacob, H.S. Exposure of endothelial cells to free heme potentiates damage mediated by granulocytes and toxic oxygen species. Lab. Investig. 1991, 64, 648–655.
- Okubo, K.; Kurosawa, M.; Kamiya, M.; Urano, Y.; Suzuki, A.; Yamamoto, K.; Hase, K.; Homma, K.; Sasaki, J.; Miyauchi, H.; et al. Macrophage extracellular trap formation promoted by platelet activation is a key mediator of rhabdomyolysis-induced acute kidney injury. Nat. Med. 2018, 24, 232–238.
- Boudhabhay, I.; Poillerat, V.; Grunenwald, A.; Torset, C.; Leon, J.; Daugan, M.V.; Lucibello, F.; El Karoui, K.; Ydee, A.; Chauvet, S.; et al. Complement activation is a crucial driver of acute kidney injury in rhabdomyolysis. Kidney Int. 2021, 99, 581–597.
- Arruda, M.A.; Rossi, A.G.; de Freitas, M.S.; Barja-Fidalgo, C.; Graca-Souza, A.V. Heme inhibits human neutrophil apoptosis: Involvement of phosphoinositide 3-kinase, MAPK, and NF-kappaB. J. Immunol. 2004, 173, 2023–2030.
- Gbotosho, O.T.; Kapetanaki, M.G.; Kato, G.J. The worst things in life are free: The role of free heme in sickle cell disease. Front. Immunol. 2020, 11, 561917.
- Poss, K.D.; Tonegawa, S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc. Natl. Acad. Sci. USA 1997, 94, 10919–10924.
- Yachie, A.; Niida, Y.; Wada, T.; Igarashi, N.; Kaneda, H.; Toma, T.; Ohta, K.; Kasahara, Y.; Koizumi, S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J. Clin. Investig. 1999, 103, 129–135.
- Yachie, A. Heme oxygenase-1 deficiency and oxidative stress: A review of 9 independent human cases and animal models. Int. J. Mol. Sci. 2021, 22, 1514.
- Sun, J.; Hoshino, H.; Takaku, K.; Nakajima, O.; Muto, A.; Suzuki, H.; Tashiro, S.; Takahashi, S.; Shibahara, S.; Alam, J.; et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002, 21, 5216–5224.
- Lin, Q.; Weis, S.; Yang, G.; Weng, Y.H.; Helston, R.; Rish, K.; Smith, A.; Bordner, J.; Polte, T.; Gaunitz, F.; et al. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J. Biol. Chem. 2007, 282, 20621–20633.
- Kovtunovych, G.; Eckhaus, M.A.; Ghosh, M.C.; Ollivierre-Wilson, H.; Rouault, T.A. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: Effects on macrophage viability and tissue iron distribution. Blood 2010, 116, 6054–6062.
- Tzima, S.; Victoratos, P.; Kranidioti, K.; Alexiou, M.; Kollias, G. Myeloid heme oxygenase-1 regulates innate immunity and autoimmunity by modulating IFN-beta production. J. Exp. Med. 2009, 206, 1167–1179.
- Gullotta, F.; di Masi, A.; Ascenzi, P. Carbon monoxide: An unusual drug. IUBMB Life 2012, 64, 378–386.
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650.
- Lee, S.; Lee, S.J.; Coronata, A.A.; Fredenburgh, L.E.; Chung, S.W.; Perrella, M.A.; Nakahira, K.; Ryter, S.W.; Choi, A.M. Carbon monoxide confers protection in sepsis by enhancing beclin 1-dependent autophagy and phagocytosis. Antioxid. Redox Signal. 2014, 20, 432–442.
- Jung, S.S.; Moon, J.S.; Xu, J.F.; Ifedigbo, E.; Ryter, S.W.; Choi, A.M.; Nakahira, K. Carbon monoxide negatively regulates NLRP3 inflammasome activation in macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L1058–L1067.
- Pecorella, S.R.; Potter, J.V.; Cherry, A.D.; Peacher, D.F.; Welty-Wolf, K.E.; Moon, R.E.; Piantadosi, C.A.; Suliman, H.B. The HO-1/CO system regulates mitochondrial-capillary density relationships in human skeletal muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L857–L871.
- Ryter, S.W. Heme oxygenase-1/carbon monoxide as modulators of autophagy and inflammation. Arch. Biochem. Biophys. 2019, 678, 108186.
- Liu, Y.; Liu, J.; Tetzlaff, W.; Paty, D.W.; Cynader, M.S. Biliverdin reductase, a major physiologic cytoprotectant, suppresses experimental autoimmune encephalomyelitis. Free Radic. Biol. Med. 2006, 40, 960–967.
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046.
- Ryter, S.W. Bile pigments in pulmonary and vascular disease. Front. Pharmacol. 2012, 3, 39.
- Ollinger, R.; Wang, H.; Yamashita, K.; Wegiel, B.; Thomas, M.; Margreiter, R.; Bach, F.H. Therapeutic applications of bilirubin and biliverdin in transplantation. Antioxid. Redox Signal. 2007, 9, 2175–2185.
- Arosio, P.; Ingrassia, R.; Cavadini, P. Ferritins: A family of molecules for iron storage, antioxidation and more. Biochim. Biophys. Acta 2009, 1790, 589–599.
- Ferreira, C.; Bucchini, D.; Martin, M.E.; Levi, S.; Arosio, P.; Grandchamp, B.; Beaumont, C. Early embryonic lethality of H ferritin gene deletion in mice. J. Biol. Chem. 2000, 275, 3021–3024.
- Zarjou, A.; Black, L.M.; McCullough, K.R.; Hull, T.D.; Esman, S.K.; Boddu, R.; Varambally, S.; Chandrashekar, D.S.; Feng, W.; Arosio, P.; et al. Ferritin light chain confers protection against sepsis-induced inflammation and organ injury. Front. Immunol. 2019, 10, 131.
- Sikura, K.E.; Potor, L.; Szerafin, T.; Zarjou, A.; Agarwal, A.; Arosio, P.; Poli, M.; Hendrik, Z.; Mehes, G.; Oros, M.; et al. Potential role of H-ferritin in mitigating valvular mineralization. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 413–431.
- Zarjou, A.; Jeney, V.; Arosio, P.; Poli, M.; Antal-Szalmas, P.; Agarwal, A.; Balla, G.; Balla, J. Ferritin prevents calcification and osteoblastic differentiation of vascular smooth muscle cells. J. Am. Soc. Nephrol. 2009, 20, 1254–1263.
- Zarjou, A.; Bolisetty, S.; Joseph, R.; Traylor, A.; Apostolov, E.O.; Arosio, P.; Balla, J.; Verlander, J.; Darshan, D.; Kuhn, L.C.; et al. Proximal tubule H-ferritin mediates iron trafficking in acute kidney injury. J. Clin. Investig. 2013, 123, 4423–4434.
- Bolisetty, S.; Zarjou, A.; Hull, T.D.; Traylor, A.M.; Perianayagam, A.; Joseph, R.; Kamal, A.I.; Arosio, P.; Soares, M.P.; Jeney, V.; et al. Macrophage and epithelial cell H-ferritin expression regulates renal inflammation. Kidney Int. 2015, 88, 95–108.
- Zarjou, A.; Kim, J.; Traylor, A.M.; Sanders, P.W.; Balla, J.; Agarwal, A.; Curtis, L.M. Paracrine effects of mesenchymal stem cells in cisplatin-induced renal injury require heme oxygenase-1. Am. J. Physiol. Renal Physiol. 2011, 300, F254–F262.
- Bolisetty, S.; Traylor, A.; Joseph, R.; Zarjou, A.; Agarwal, A. Proximal tubule-targeted heme oxygenase-1 in cisplatin-induced acute kidney injury. Am. J. Physiol. Renal Physiol. 2016, 310, F385–F94.
- Nath, K.A.; Haggard, J.J.; Croatt, A.J.; Grande, J.P.; Poss, K.D.; Alam, J. The indispensability of heme oxygenase-1 in protecting against acute heme protein-induced toxicity in vivo. Am. J. Pathol. 2000, 156, 1527–1535.
- Kim, J.; Zarjou, A.; Traylor, A.M.; Bolisetty, S.; Jaimes, E.A.; Hull, T.D.; George, J.F.; Mikhail, F.M.; Agarwal, A. In vivo regulation of the heme oxygenase-1 gene in humanized transgenic mice. Kidney Int. 2012, 82, 278–291.
- Wei, Q.; Hill, W.D.; Su, Y.; Huang, S.; Dong, Z. Heme oxygenase-1 induction contributes to renoprotection by G-CSF during rhabdomyolysis-associated acute kidney injury. Am. J. Physiol. Renal Physiol. 2011, 301, F162–F170.
- Boddu, R.; Hull, T.D.; Bolisetty, S.; Hu, X.; Moehle, M.S.; Daher, J.P.; Kamal, A.I.; Joseph, R.; George, J.F.; Agarwal, A.; et al. Leucine-rich repeat kinase 2 deficiency is protective in rhabdomyolysis-induced kidney injury. Hum. Mol. Genet. 2015, 24, 4078–4093.
- Wu, J.; Pan, X.; Fu, H.; Zheng, Y.; Dai, Y.; Yin, Y.; Chen, Q.; Hao, Q.; Bao, D.; Hou, D. Effect of curcumin on glycerol-induced acute kidney injury in rats. Sci. Rep. 2017, 7, 10114.
- Shah, S.V.; Walker, P.D. Evidence suggesting a role for hydroxyl radical in glycerol-induced acute renal failure. Am. J. Physiol. 1988, 255, F438–F443.
- Paller, M.S. Hemoglobin- and myoglobin-induced acute renal failure in rats: Role of iron in nephrotoxicity. Am. J. Physiol. 1988, 255, F539–F544.
- Pamplona, A.; Ferreira, A.; Balla, J.; Jeney, V.; Balla, G.; Epiphanio, S.; Chora, A.; Rodrigues, C.D.; Gregoire, I.P.; Cunha-Rodrigues, M.; et al. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat. Med. 2007, 13, 703–710.
- Pereira, M.L.; Ortolan, L.S.; Sercundes, M.K.; Debone, D.; Murillo, O.; Lima, F.A.; Marinho, C.R.; Epiphanio, S. Association of heme oxygenase 1 with lung protection in malaria-associated ALI/ARDS. Mediat. Inflamm. 2016, 2016, 4158698.
- Aubouy, A.; Olagnier, D.; Bertin, G.; Ezinmegnon, S.; Majorel, C.; Mimar, S.; Massougbodji, A.; Deloron, P.; Pipy, B.; Coste, A. Nrf2-driven CD36 and HO-1 gene expression in circulating monocytes correlates with favourable clinical outcome in pregnancy-associated malaria. Malar. J. 2015, 14, 358.
- Ramos, S.; Carlos, A.R.; Sundaram, B.; Jeney, V.; Ribeiro, A.; Gozzelino, R.; Bank, C.; Gjini, E.; Braza, F.; Martins, R.; et al. Renal control of disease tolerance to malaria. Proc. Natl. Acad. Sci. USA 2019, 116, 5681–5686.
- Nath, K.A.; Hebbel, R.P. Sickle cell disease: Renal manifestations and mechanisms. Nat. Rev. Nephrol. 2015, 11, 161–171.
- Muller-Eberhard, U.; Javid, J.; Liem, H.H.; Hanstein, A.; Hanna, M. Plasma concentrations of hemopexin, haptoglobin and heme in patients with various hemolytic diseases. Blood 1968, 32, 811–815.
- Saraf, S.L.; Zhang, X.; Shah, B.; Kanias, T.; Gudehithlu, K.P.; Kittles, R.; Machado, R.F.; Arruda, J.A.; Gladwin, M.T.; Singh, A.K.; et al. Genetic variants and cell-free hemoglobin processing in sickle cell nephropathy. Haematologica 2015, 100, 1275–1284.
- Nath, K.A.; Grande, J.P.; Haggard, J.J.; Croatt, A.J.; Katusic, Z.S.; Solovey, A.; Hebbel, R.P. Oxidative stress and induction of heme oxygenase-1 in the kidney in sickle cell disease. Am. J. Pathol. 2001, 158, 893–903.
- Ghosh, S.; Tan, F.; Yu, T.; Li, Y.; Adisa, O.; Mosunjac, M.; Ofori-Acquah, S.F. Global gene expression profiling of endothelium exposed to heme reveals an organ-specific induction of cytoprotective enzymes in sickle cell disease. PLoS ONE 2011, 6, e18399.
- Camus, S.M.; De Moraes, J.A.; Bonnin, P.; Abbyad, P.; Le Jeune, S.; Lionnet, F.; Loufrani, L.; Grimaud, L.; Lambry, J.C.; Charue, D.; et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood 2015, 125, 3805–3814.
- Belcher, J.D.; Mahaseth, H.; Welch, T.E.; Otterbein, L.E.; Hebbel, R.P.; Vercellotti, G.M. Heme oxygenase-1 is a modulator of inflammation and vaso-occlusion in transgenic sickle mice. J. Clin. Investig. 2006, 116, 808–816.
- Nath, K.A.; Katusic, Z.S. Vasculature and kidney complications in sickle cell disease. J. Am. Soc. Nephrol. 2012, 23, 781–784.
- Payan-Pernia, S.; Ruiz Llobet, A.; Remacha Sevilla, A.F.; Egido, J.; Ballarin Castan, J.A.; Moreno, J.A. Sickle cell nephropathy. Clinical manifestations and new mechanisms involved in kidney injury. Nefrologia 2021.
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O., 3rd; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 2002, 8, 1383–1389.
- Taha, H.; Skrzypek, K.; Guevara, I.; Nigisch, A.; Mustafa, S.; Grochot-Przeczek, A.; Ferdek, P.; Was, H.; Kotlinowski, J.; Kozakowska, M.; et al. Role of heme oxygenase-1 in human endothelial cells: Lesson from the promoter allelic variants. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1634–1641.
- Johnson, A.C.; Becker, K.; Zager, R.A. Parenteral iron formulations differentially affect MCP-1, HO-1, and NGAL gene expression and renal responses to injury. Am. J. Physiol. Renal Physiol. 2010, 299, F426–F435.
- Leaf, D.E.; Rajapurkar, M.; Lele, S.S.; Mukhopadhyay, B.; Rawn, J.D.; Frendl, G.; Waikar, S.S. Increased plasma catalytic iron in patients may mediate acute kidney injury and death following cardiac surgery. Kidney Int. 2015, 87, 1046–1054.
- Leaf, D.E.; Rajapurkar, M.; Lele, S.S.; Mukhopadhyay, B.; Waikar, S.S. Plasma catalytic iron, AKI, and death among critically ill patients. Clin. J. Am. Soc. Nephrol. 2014, 9, 1849–1856.
- Hatcher, H.C.; Tesfay, L.; Torti, S.V.; Torti, F.M. Cytoprotective Effect of Ferritin H in Renal Ischemia Reperfusion Injury. PLoS ONE 2015, 10, e0138505.
- Scindia, Y.; Dey, P.; Thirunagari, A.; Liping, H.; Rosin, D.L.; Floris, M.; Okusa, M.D.; Swaminathan, S. Hepcidin mitigates renal ischemia-reperfusion injury by modulating systemic iron homeostasis. J. Am. Soc. Nephrol. 2015, 26, 2800–2814.
- Van Swelm, R.P.; Wetzels, J.F.; Verweij, V.G.; Laarakkers, C.M.; Pertijs, J.C.; van der Wijst, J.; Thevenod, F.; Masereeuw, R.; Swinkels, D.W. Renal handling of circulating and renal-synthesized hepcidin and its protective effects against hemoglobin-mediated kidney injury. J. Am. Soc. Nephrol. 2016, 27, 2720–2732.


