Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sandra Casimiro | + 4238 word(s) | 4238 | 2021-08-10 08:18:25 | | | |
| 2 | Rita Xu | Meta information modification | 4238 | 2021-08-10 11:28:54 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Casimiro, S. RANKL/RANK Pathway in Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/13003 (accessed on 26 June 2026).
Casimiro S. RANKL/RANK Pathway in Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/13003. Accessed June 26, 2026.
Casimiro, Sandra. "RANKL/RANK Pathway in Cancer" Encyclopedia, https://encyclopedia.pub/entry/13003 (accessed June 26, 2026).
Casimiro, S. (2021, August 10). RANKL/RANK Pathway in Cancer. In Encyclopedia. https://encyclopedia.pub/entry/13003
Casimiro, Sandra. "RANKL/RANK Pathway in Cancer." Encyclopedia. Web. 10 August, 2021.
Copy Citation
The receptor activator of the nuclear factor-κB ligand (RANKL)/RANK signaling pathway was identified in the late 1990s and is the key mediator of bone remodeling. Targeting RANKL with the antibody denosumab is part of the standard of care for bone loss diseases, including bone metastases (BM). During the past decade RANKL/RANK pathway has emerged as an important player in breast carcinogenesis and response to immunotherapy in different solid tumors, gaining a new insight into the pan-cancer benefits of targeting of the RANKL/RANK pathway in cancer.
bone metastasis
bone-targeted agent
breast cancer
drug repurposing
RANK ligand (RANKL)
1. Introduction
Identified over two decades ago [1][2][3], the receptor activator of the nuclear factor-κB ligand (RANKL)/RANK pathway remains a hot topic in cancer research. Long studied for its role as a master regulator of osteoclastogenesis [1][4], this pathway gained renewed interest over the past decade as an important player in breast carcinogenesis [5][6][7]. More recently, compelling evidence supporting the role of the RANKL/RANK pathway in response to immunotherapy [8][9][10] fueled translational and clinical research on the biology and targeting of the RANKL/RANK pathway in cancer.
RANKL (also known as TRANCE) is a homotrimeric transmembrane protein member of the tumor necrosis factor (TNF) cytokine family, initially identified as a mediator of T cell-dependent immune response, particularly in the modulation of T cell [11] and dendritic cell (DC) activity [12]. However, it was soon found to be the same molecule as the so-called osteoclast differentiation factor (ODF), which was already known to promote functional osteoclast differentiation in presence of colony-stimulating factor 1 (CSF-1) [2][4]. Soluble RANKL derives from proteolytic cleavage by matrix metalloproteinase (MMP)-14 and A disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) [13] or alternative splicing [14]. RANK is a transmembrane receptor, member of the TNF receptor (TNFR) family, activated by RANKL binding. Expressed on the surface of osteoclasts, RANK’s physiological role in bone was confirmed in vivo by the observation that RANK-deficient mice suffered from severe osteopetrosis because of impaired osteoclast differentiation [1]. The RANKL/RANK signaling axis involves a third player, osteoprotegerin (OPG), a soluble decoy receptor with a high affinity for RANKL, which is critical for bone homeostasis [15][16].
Due to its pivotal role in bone pathophysiology, efforts have been made regarding the pharmacological targeting of the RANKL/RANK pathway as a way to prevent bone resorption, with the first attempts going back to the early 2000s through the use of recombinant OPG (OPG-Fc) [17]. However, OPG-Fc clinical development was discontinued in favor of denosumab, a fully human immunoglobulin G (IgG) 2 monoclonal antibody that binds specifically to RANKL, which was later approved for the treatment of osteoporosis and cancer-induced bone metastases (BM) [18][19]. Denosumab is a very interesting example of drug repurposing, since this bone-targeted agent (BTA) is currently under investigation in other clinical settings, with promising results.
This entry provides a parallel perspective of the pre-clinical and clinical evidence of RANKL/RANK pathway in cancer-induced BM, breast cancer (BCa) onset and progression, and immune modulation (Figure 1), exploring the (potential) efficacy of RANKL inhibition in all cancer stages.
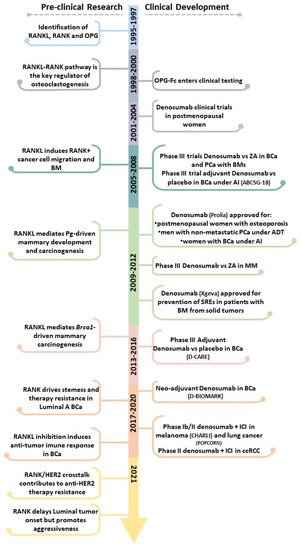
Figure 1. Pre-clinical and clinical landmarks of RANKL/RANK pathway research in Oncology. ADT, androgen deprivation therapy; AI, aromatase inhibitors; BCa, breast cancer; BM, bone metastases; ccRCC, clear cell renal cell carcinoma; ICI, immune checkpoint inhibitor; MM, multiple myeloma; PCa, prostate cancer; Pg, progesterone; SREs, skeletal-related effects; ZA, Zoledronate.
2. The Role of RANKL/RANK Pathway in Bone Health and Disease
BM represent the most common form of distant relapse in malignancies with high incidence, as BCa and prostate cancer (PCa), and are highly prevalent in renal cell carcinoma (RCC) and multiple myeloma (MM). However, several other cancers metastasize to the skeleton. With very specific biology and pathophysiology, strictly related to unbalanced bone resorption, BM are associated with high morbidity [20][21]. BM tumor cells growing at bone metastatic niche totally subvert the physiologically balanced bone homeostasis to favor cancer spread, leading to osteolysis, skeletal-related events (SREs), and decreased overall survival (OS).
Osteoclasts and osteoblasts are the bone cells responsible for bone resorption and formation, respectively. Physiologic and pathologic osteoclastogenesis is triggered by RANKL produced by bone marrow stromal cells, osteocytes, and osteoblasts, which binds to RANK on the surface of hematopoietic osteoclast precursor cells [22]. This leads to recruitment of TNFR-associated cytoplasmic factors (TRAFs) for specific cytoplasmic RANK domains, subsequent activation of NF-κB, c-Jun N-terminal kinase (JNK), p38, extracellular signal-regulated kinase (ERK), and Src pathways, and expression of genes characteristic of active, bone-resorbing osteoclasts. CSF-1, interleukin (IL)-6, IL-8, and chemokine (C-X-C motif) ligand 12 (CXCL2) are also important regulators of osteoclastogenesis. In turn, osteoclast-mediated bone resorption releases osteoblastic factors from the bone matrix, including transforming growth factor-beta (TGF-β), bone morphogenetic proteins (BMPs), fibroblast growth factor (FGF), platelet-derived growth factor (PDGF), and insulin-like growth factors (IGFs), inducing osteoblast differentiation from stromal mesenchymal stem cells. Subsequently, OPG secreted by mature osteoblasts and stromal cells exerts a negative effect over osteoclastogenesis by sequestering RANKL.
However, metastatic tumor cells in bone express a panoply of osteoclastogenic factors that drive increased bone resorption. These include IL-1, IL-6, parathyroid hormone-related protein (PTHrP), prostaglandin E2 (PEG2), CSF-1, and TNF-alpha (TNF-α). Tumor-derived PTHrP and bone-derived IL-11 upregulate RANKL and downregulate OPG, thereby activating osteoclastogenesis. In response, exacerbated bone resorption feeds tumor cells with mitogenic factors released from the bone matrix, like TGF-β, IGFs, FGFs, PDGF, and Ca2+, further fueling the so-called “vicious cycle of BM” [20][23][24][25].
Although all BM present increased bone resorption, osteoblastic BM, as seen in PCa, also display augmented bone formation. This mostly occurs via cancer cell-produced endothelin-1 (ET-1), which stimulates endothelin A receptor(ETR) in osteoblasts, activating Wnt signaling and osteoblast activity [26].
The efficacy of RANKL inhibition in the treatment of BM was demonstrated in different in vivo models of breast, lung, prostate, and colon cancer [19][27]. Ultimately, indifferent to the radiologic nature of BM or tumor type, RANKL/RANK pathway blockade is able to abrogate cancer-induced osteoclastogenesis and bone resorption, delaying and decreasing SREs [28]. The clinical benefit of RANKL pharmacological inhibition in BM will be discussed in the next section. Yet, it is important to review the molecular and biological effects of RANKL blockade, along with evidence that rationalizes the clinical benefit of denosumab over other BTAs, like bisphosphonates (BPs). In the next sections, the impact of RANKL blockade in osteoclasts’ life span, (in)direct anti-tumor effects, and bone pre-metastatic niche modulation will be explored (Figure 2).
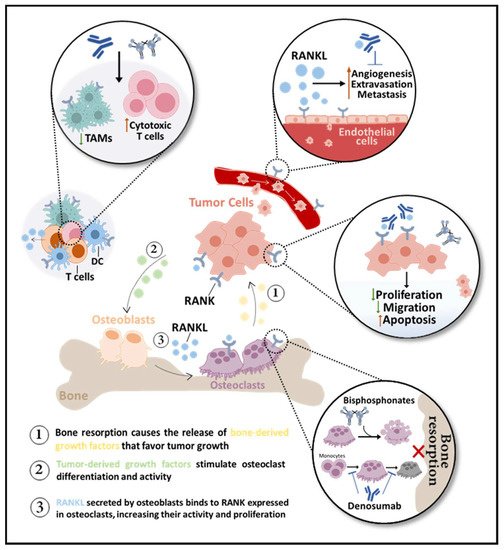
Figure 2. RANKL inhibition in bone metastases (BM). Bone-targeted agents (BTAs) are used to control BM by impairing bone resorption, indirectly affecting the tumor burden. While bisphosphonates only affect mature osteoclasts, inducing apoptosis, the anti-RANKL antibody denosumab prevents osteoclast differentiation, activity, and survival. Denosumab may also be antiangiogenic over RANK-positive endothelial cells. RANK is expressed in dendritic cells (DC) and macrophages, like tumor-associated macrophages (TAMs), and T-cell derived RANKL inhibition decreases TAMs and increases cytotoxic T cells. Potential direct effects of BTAs in tumor cells include inhibition of proliferation and migration and induction of apoptosis.
2.1. Osteoclasts
Signal transduction through RANK not only drives osteoclast differentiation from hematopoietic precursors, but also activates and prolongs the survival of mature osteoclasts [29][30]. Formation of RANKL-induced actin ring in mature osteoclasts occurs within minutes of RANK activation, and RANKL-treated mice show increased blood ionized Ca2+ within one hour after injection, consistently with immediate osteoclast activation in vivo [29]. Additionally, RANKL (through NFkB/JNK/Src) and CSF-1 (through NFkB/bcl-2) are required for optimal osteoclast survival [30].
Conversely, BPs—structural analogs of pyrophosphates with high affinity to hydroxyapatite in bone—are only effective against bone-resorbing mature osteoclasts, upon internalization [31]. BPs induce osteoclast apoptosis either by causing intracellular accumulation of adenosine triphosphate (ATP) toxic analogs (non-N-BPs, like etidronate and clodronate) or by interfering with farnesyl pyrophosphate synthase (FPPS)—a key enzyme in the mevalonate pathway and protein prenylation (N-BPs, like pamidronate, alendronate, and zoledronate [ZA]).
Therefore, blocking RANKL/RANK pathway controls pre-osteoclast recruitment, fusion into multinucleated osteoclasts, osteoclast activation, and osteoclast survival, and this may render an advantage over BPs, which are effective in impairing the activity of mature osteoclasts but not in preventing their differentiation. This influences bone resorption rate, as evidenced by the significant decrease in urinary NTx/creatinine levels—a biomarker of bone resorption—with denosumab compared with ZA in patients with BM, independently of tumor type [32][33][34].
2.2. (In)Direct Anti-Tumor Effects
It is widely accepted that BTAs have an indirect anticancer effect, by decreasing bone resorption and consequently reducing the availability of matrix-trapped mitogens to fuel tumor growth. However, there is compelling pre-clinical evidence that BTAs may also directly affect metastatic tumor cells. As denosumab does not recognize murine RANKL, the vast majority of pre-clinical studies use OPG-Fc (and RANK-Fc) to mimic denosumab.
In vitro preclinical evidence from diverse tumor types—including BCa, PCa, lung, ovarian, and bladder cancer, and RCC—suggests that BPs have direct effects over tumor cells, namely through apoptosis induction and proliferation, migration, and invasion inhibition [35][36]. Moreover, several cancer cells, including osteotropic BCa and PCa ones, express functional RANK, which induces downstream pathway activation upon RANKL stimulus, increasing migration and invasion [37][38][39][40][41], endorsing a role for RANK signaling in the acquisition of a more aggressive phenotype. However, one study in a murine BCa BM model using the osteotropic MDA-MB-231 cell line directly compared the use of ibandronate with OPG-Fc with therapeutic intents and showed no efficacy differences between either one or the combination of both [42], advocating only indirect effects of both BTAs on tumor growth. Nonetheless, RANK overexpression in the same cell line significantly increased metastatic growth rate in bone versus parental cells and, although RANKL inhibition and ZA reduced BM, RANKL inhibition was more effective, suggesting a direct effect over RANK-positive (RANK+) tumors [43]. The observation that RANKL upregulates osteotropic gene expression in cancer cells, favoring osteoclastogenesis, supported this hypothesis. Additionally, our group demonstrated that RANKL is a positive regulator of MMP-1 in MDA-MB-231 cells [39], a known osteotropic factor [44][45]. Moreover, OPG-Fc prevented tumor-induced BM in a mouse model of estrogen receptor-positive (ER+) BCa, where tamoxifen as a single agent was shown to reduce tumor growth in the hind limbs and OPG-Fc blocked bone resorption, but the combination of both was more effective [46]. The same study hypothesized that RANKL inhibition targeted the bone microenvironment and tamoxifen cancer cells. However, hawse have recently shown that RANK overexpression in ER+ BCa cells is associated with endocrine therapy (ET) resistance [47], indicating that RANKL inhibition may have increased sensitivity to tamoxifen according to results of the previous study.
In PCa, pre-clinical studies have also come to different conclusions, supporting both the “indirect-only” and “direct” anti-tumor effect hypotheses. In the LNCaP PCa mouse model, OPG-Fc had no effect in cell viability, proliferation, or basal apoptotic rate in vitro or in vivo in subcutaneous tumors, despite being effective in preventing BM development [48]. The same inefficacy in subcutaneous tumors or in vitro proliferation was reported in other studies with LNCaP [49] and LuCaP [50][51] cell lines. In vitro, RANKL expressed by LNCaP cells was shown to be osteoclastogenic [48]. It was recently reported that soluble RANKL is dispensable for physiological regulation of bone and immune systems or non-skeletal metastases, but seems to have an important role in promoting BM development by directly triggering migration of tumor cells to bone [52]. This additional evidence supports the preventive role of RANKL targeting through direct osteotropism inhibition. Accordingly, RANKL was able to trigger LNCaP [37] and PC3 [38] PCa cell migration in vitro.
In favor of the “direct” anti-tumor effect, a recent study addressing the predictive value of RANK+ circulating cancer cells (CTCs) in metastatic BCa patients during denosumab treatment demonstrated that 70% of patients with detectable CTCs had one or more RANK+ CTCs [53]. Interestingly, whereas total baseline CTCs were associated with bone outcomes, RANK+ CTC persistence during treatment correlated with better outcomes, suggesting its relevance in RANKL inhibition efficacy.
Overall, the direct anti-tumor effect of RANKL inhibition seems to be more deleterious to the metastatic properties of cancer cells than BPs, further contributing to the clinical benefit of BM treatment, which will be further discussed below.
2.3. Bone Pre-Metastatic Niche
Apart from RANKL effects on osteoclasts and tumor cells, the role of RANKL targeting in the tumor microenvironment is also potentially relevant, extending its effects on BM development to BM prevention. RANKL was found to be an inductor of angiogenesis and increased vascular permeability in RANK-expressing endothelial cells, which may favor extravasation and metastases [54]. In this case, RANKL inhibition may also have an antiangiogenic effect, decreasing relapse in distant organs. However, the rationale for using anti-RANKL or other BTAs in adjuvant settings to prevent bone relapse relies on a decrease in bone resorption and bone-derived chemoattractant molecules, as well as on making bone a less congenial soil for cancer cell growth.
It is acknowledged that osteotropic cancer cells use CXCR4 to “sense” CXCL12 at distant locations, including bone [55]. In addition, evidence indicates that tumor cells at the primary location can modulate the bone microenvironment as a pre-metastatic niche [56][57]. Examples include increased MAF-regulated PTHrP expression [58] and expression of the macrophage-capping protein (CAPG) and PDZ domain-containing protein GIPC1 (GIPC1) [59] in bone-tropic primary BCa. Moreover, exosomes released from PCa cells relate with BM incidence and may modulate the pre-metastatic niche [60]. Therefore, in a preventive setting, targeting both primary cancer cells and bone microenvironment may affect BM onset.
Pre-clinical murine studies addressing RANKL inhibition in both preventive and therapeutic settings have shown that targeting RANK-expressing cancer cells not only decreases BM tumor burden but also prevents BM onset [37][61]. This suggests the combination of a less favorable pre-metastatic niche—depriving tumor cells of growth factors and cytokines that would be released from bone—and decreased cancer cell metastatic, or at least osteotropic, characteristics. The preventive effect of RANKL inhibition in BM was found to be dependent on the direct effect in RANK-mediated expression of cancer cell-derived osteotropic factors [37][38][39] and also to be more effective than BPs in tumors with high RANK expression [43].
According to the pathophysiology of BM, a highly resorptive post-menopausal bone would be a more attractive pre-metastatic niche for cancer cells to thrive. However, analysis of clinical series looking at disseminated tumors cells in bone marrow suggests that the longer a woman is post-menopausal, the less attractive bone microenvironment is for tumor cells [62][63]. A recent study has shown that estradiol and TGF-β upregulate PTHrP in ER+ osteotropic BCa cells [64] and increase the progression of osteolytic metastases in ER-negative BCa cells [65]. Therefore, estrogen deficiency may contribute to the benefit of adjuvant BPs in bone recurrence in post-menopausal women, while in metastatic settings, the menopausal status does not affect outcomes related to BTAs [66]. In agreement with these data, ZA reduced BM in oophorectomized mice in an animal model of BCa-induced BM [67]. However, in adjuvant setting RANKL inhibition reduced disease recurrence in bone irrespectively of menopausal status [68], which may be linked to the crosstalk between estrogen and RANKL. Estrogen is a known regulator of bone physiology and pathophysiology and has been shown to protect bone by regulating osteoclast survival, either by inducing autocrine TGF-β expression or by upregulating the apoptosis-promoting Fas ligand in osteoblasts [69][70]. Upon estrogen withdrawal, increased RANKL expression is the main mechanism underlying bone turnover upregulation. Therefore, lower RANKL levels in pre-menopausal conditions may contribute to the efficacy of RANKL inhibition. The clinical outcomes of BTAs in BM and adjuvant settings will be presented later in this review.
Recently, it has been hypothesized that the effects of BTAs in the bone microenvironment immune compartment may significantly account for pre-metastatic niche modulation [62]. BPs were shown to reduce CD11b+ tumor-associated macrophages (TAMs), decreasing vascular endothelial growth factor (VEGF) and MMP-9 in the tumor microenvironment and leading to anti-angiogenesis [71]. TAM reduction accompanies TAM repolarization into anti-tumor M1 macrophages [72]. Moreover, BPs can also activate cytotoxic γδT cells (Vγ9Vδ2) and promote their infiltration into tumors, which will sense IPP/ApppI-BP-induced accumulation in cancer cells as phosphoantigens [73][74]. These immunomodulatory effects of BPs have been suggested as a reason for their advantage over RANKL inhibition in preventing BM in the adjuvant setting [75].
Although RANKL/RANK pathway is an important immune regulator and studies have hypothesized that RANKL inhibition in patients with BM could be immunosuppressive, clinically significant effects on the immune system have not been reported in clinical trials. Evidence suggests that RANKL is an effective, but not essential, co-stimulatory factor for immune cell activation, supporting the lack of relevant clinical findings regarding the impact of RANKL inhibition in immunity. It was shown that the RANKL effect in T cells is redundant with other cytokines, and that only when major immunologically active molecules are deleted does RANKL/RANK pathway become the main co-stimulatory pathway for crosstalk between immune cells [76]. For example, although monocyte−macrophages are positively regulated by RANKL (protected from apoptosis, increased phagocytic properties, and activated antigen presentation), RANKL−/− mice have no alteration in the number and distribution of monocyte–macrophages [1], except if co-stimulatory molecules are missing (e.g., CD40L) [76]. Moreover, RANKL enhances DC survival, antigen presentation, and cytokine production in vitro, but RANK and RANKL−/− mice have intact DC development and function [1].
Overall, the complex crosstalk between cancer cells, bone pre-metastatic niche, and tumor microenvironment is clearly affected by RANK expression outside osteoclasts, and RANKL blockade may contribute to improved outcomes in BM patients through several complementary mechanisms. These include the indirect impact on tumor growth through a decrease in bone soil congeniality; the indirect impact mediated by anti-angiogenic properties; the direct impact in cell signaling, migration, invasion, and osteotropism; and the decreased ability of cancer cells to prepare the pre-metastatic niche in the bone. In the next section, clinical evidence on RANKL inhibition in BM will be summarized.
2.4. Anti-RANKL Therapy in BM: Discovery and Current and Future Perspectives
The anti-resorptive effect of recombinant human OPG was reported two decades ago, after observations that its intravenous administration in normal rats increased bone mineral density and bone volume as a consequence of decreased active osteoclasts [17]. This was in accordance with observations that OPG-deficient mice developed early osteopenia [77]. However, despite its unequivocal physiological ability to impair bone resorption, very high subcutaneous doses (>10–30 mg/kg) of recombinant full-length OPG were required for in vivo efficacy, and its pharmacokinetic and pharmacodynamic profile was poor [78]. The best protein was a recombinant protein comprising the a.a. 22–194 of human OPG fused with the human IgG1 Fc region, found to be over 200 times more active than full-length OPG in vivo and with prolonged half-life. Nonetheless, OPG-Fc and RANK-Fc were associated with autoimmune hypercalcemia, being discontinued in favor of an anti-RANK antibody and ultimately leading to AMG 16, currently known as denosumab [19].
2.4.1. Bone Metastatic Disease
Although the treatment of BM from solid tumors and MM is rarely curative, it is possible to prevent disease progression and palliate symptoms for many years using systemic anticancer treatments [20]. SREs reflect the burden of bone pain and structural damage caused by bone metastatic involvement, representing an important form of skeletal morbidity that impacts patients’ quality of life and results in significant healthcare costs [79]. SREs comprise five major complications of tumor bone disease: pathological fracture, need for radiotherapy to relieve bone pain or reduce bone structural damage, need for bone surgery to prevent or repair fractures, spinal cord compression, and hypercalcemia [20]. BTAs have been shown to improve bone structure and quality, minimizing the risk of SREs in patients with BM from solid tumors and MM [75][79]. Therefore, in order to reduce morbidity and complement other cancer-specific treatments, current clinical guidelines recommend prescribing a BTA following the initial radiological diagnosis of BM in most patients [80].
To understand the current clinical applications of RANKL inhibition in the context of BM, it is important to briefly review the role of BPs in the history of bone metastatic disease management. Several BPs have proven efficacious in preventing SREs in patients with BM from BCa or MM since the approval of clodronate in these indications in the early 1990s [33][81]. Still, ZA remains the only BP approved for the treatment of metastatic castration-resistant PCa (CRPC) and BM from other solid tumors [79]. The addition of a BTA in the treatment of endocrine-sensitive PCa showed no evidence of a survival improvement or SRE reduction compared with placebo and is hence not recommended outside treatment-induced bone loss prevention or pre-existing osteoporosis clinical settings [80][82].
Since denosumab was first licensed for the treatment of BM from solid tumors in 2010, numerous head-to-head randomized controlled trials (RCTs) have compared denosumab with ZA in bone health settings in several human cancers (Table 1) [79].
Table 1. Head-to-head randomized controlled trials comparing denosumab with zoledronate for delay or prevention of skeletal-related events in bone metastatic solid tumors and multiple myeloma.
| Cancer Type(s) | First On-Study SREs (% of Patients; D vs. ZA) |
Time to First SRE | Time to First and Subsequent SREs | Ref. |
|---|---|---|---|---|
| Breast (n = 2046) |
NE | Denosumab superior (HR 0.82; 95% CI 0.71–0.95; p < 0.001 NI; p = 0.01 S) |
Denosumab superior (RR 0.77; 95% CI 0.66–0.89; p = 0.001 S) |
[32] |
| CRPC (n = 1901) |
36 vs. 41 | Denosumab superior (HR 0.82; 95% CI 0.71–0.95; p = 0.0002 NI; p = 0.008 S) |
Denosumab superior (RR 0.82; 95% CI 0.71–0.94; p = 0.008) |
[33] |
| Solid tumors (excluding breast and prostate) and MM (n = 1779) |
NE | Denosumab non-inferior, but not statistically superior (HR 0.84; 95% CI, 0.71 to 0.98; p = 0.0007 NI; p = 0.06 S) |
Denosumab not statically superior (RR 0.90; 95% CI 0.77–1.04; p = 0.14) |
[83] |
| MM (n = 1718) |
44 vs. 45 | Denosumab non-inferior, but not statistically superior (HR 0.98; 95% CI 0.85–1.14; p = 0.01 NI) |
Denosumab not statically superior (RR 1.01; 95% CI 0.89–1.15; p = 0.84) |
[83] |
CI, Confidence Interval; CRPC, Castration-Resistant PCa; D, Denosumab; HR, Hazard Ratio; MM, Multiple Myeloma; NE, Not Evaluable; NI, Non-Inferiority; RR, Rate Ratio; S, Superiority; SREs, Skeletal-Related Events; ZA, Zoledronate.
Denosumab was shown to be superior to ZA in delaying and preventing SREs in patients with bone metastatic BCa [32] and metastatic CRPC [33]. In an RCT including patients with BM from solid tumors and MM (excluding BCa and PCa), denosumab was non-inferior to ZA, but was not superior in delaying time to first and subsequent SREs [34]. However, an ad hoc analysis of this trial excluding the MM cohort was able to demonstrate a significant advantage of denosumab in delaying SREs [84]. There was no difference regarding OS or disease progression between patients treated with denosumab or ZA in each trial individually or in a combined analysis of the three trials (Table 2).
Table 2. Randomized controlled trials of denosumab disease-modifying properties in advanced human cancer.
| Cancer Type(s) | Number of Patients | Intervention | Disease-Related Outcomes | Trial Identifier/Reference |
|---|---|---|---|---|
| Breast (advanced, all types, pre-and postmenopausal) |
2046 | Denosumab vs. ZA | Similar OS (HR 0.95; 95% CI 0.81–1.11; p = 0.49) and time to disease progression (HR 1.00; 95% CI 0.89–1.11; p = 0.93). | NCT00321464 [32] |
| CRPC | 1901 | Denosumab vs. ZA | Similar OS (HR 1.03; 95% CI 0.91–1.17; p = 0.65) and time to disease progression (HR 1.06; 95% CI 0.95–1.18; p = 0.30). | NCT00321620 [33] |
| Solid tumors (excluding breast and prostate) and MM | 1779 | Denosumab vs. ZA | Similar OS (HR 0.95; 95% CI 0.83–1.08; p = 0.43) and time to disease progression (HR 1.00; 95% CI 0.89–1.12; p = 1.00). Ad hoc analyses favored denosumab for NSCLC patients (HR 0.79; 95% CI 0.65–0.95) and ZA for MM patients (HR 2.26; 95% CI 1.13–4.50). |
NCT00330759 [34] |
| NSCLC (stage IV) |
514 | ChT + Denosumab vs. ChT | Similar OS (HR 0.96; 95% CI 0.78–1.19; p = 0.36), PFS (HR 0.99; 95% CI 0.82–1.19; p = 0.46) and ORR (30.5% vs. 29.4%; p = 0.85). | NCT02129699 (SPLENDOUR) [85] |
| MM | 1718 | Denosumab vs. ZA | Denosumab improved PFS by 10.7 months (HR, 0.82; 95% CI 0.68–0.99; p = 0.036). Similar OS (HR, 0.90; 95% CI 0.70–1.16; p = 0.41). | NCT01345019 [83] |
ChT, Chemotherapy; DFS, Disease-Free Survival; MM, Multiple Myeloma; NCT, National Clinical Trial; NSCLC, Non-Small Cell Lung Cancer; ORR, Objective Response Rate; OS, Overall Survival; PFS, Progression-Free Survival; ZA, Zoledronate.
An exploratory subgroup analysis of non-small cell lung cancer (NSCLC) patients from the phase 3 trial of denosumab versus ZA in the treatment of BM from solid tumors or MM suggested a significant OS advantage for denosumab [86]. However, in the recently published SPLENDOUR trial (NCT02129699), denosumab failed to show a measurable impact in OS, progression-free survival (PFS), or objective response rate (ORR) when added to standard first-line platinum-based doublet chemotherapy in advanced NSCLC, irrespective of the presence of BM at diagnosis or histological subtype [85].
In the case of MM, the Myeloma IX trial demonstrated that ZA has anti-myeloma effects beyond SRE prevention, evidencing a median PFS and OS improvement compared with clodronate [87]. Contrarily to the observed in NSCLC, the ad hoc subgroup analysis of MM patients from the phase 3 RCT comparing denosumab with ZA in the treatment of non-breast, non-prostate bone metastatic solid tumors and MM suggested a survival advantage for ZA over denosumab. However, since this trial was considered potentially confounded by imbalances in patient characteristics, antitumor therapies, and early withdrawals and limited by the small proportion of the MM cohort (10%) [88], a larger RCT focusing exclusively on MM patients was conducted [83]. This trial evidenced that denosumab was statically non-inferior in preventing SREs and carried a PFS but not an OS advantage compared with ZA. These results led to the approval of denosumab for the prevention of SREs in patients with MM, currently representing a particularly useful alternative in patients with renal dysfunction, a common clinical consequence in MM for which BPs may be contraindicated.
In summary, a number of factors must be considered when selecting a BTA for bone health management in solid tumors or MM bone disease, namely drug availability, route of administration, and patient preference [80]. Denosumab seems to have an advantage over other BTAs due to its efficacy, convenience, and renal health benefits. However, BPs may be more cost-effective.
Discontinuation is another important aspect to consider regarding BTAs. While BPs incorporate into the bone matrix, having a prolonged action duration, denosumab has a short half-life and bone turnover suppression is not maintained after discontinuation. This justifies that the frequency of ZA administration may be reduced during disease remission periods or even that ZA is interrupted to allow safer dental treatments without substantially influencing the risk of SREs. Denosumab discontinuation, on the other hand, can result in rebound osteolysis that may lead to rapid bone loss, increased bone pain, and increased risk of SREs [89][90]. This supports the current recommendation to use BPs after stopping denosumab, as a way to minimize the clinical consequences of this rebound phenomenon [80].
References
- Dougall, W.C.; Glaccum, M.; Charrier, K.; Rohrbach, K.; Brasel, K.; De Smedt, T.; Daro, E.; Smith, J.; Tometsko, M.E.; Maliszewski, C.R.; et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999, 13, 2412–2424.
- Yasuda, H.; Shima, N.; Nakagawa, N.; Yamaguchi, K.; Kinosaki, M.; Mochizuki, S.; Tomoyasu, A.; Yano, K.; Goto, M.; Murakami, A.; et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA 1998, 95, 3597–3602.
- Kong, Y.Y.; Yoshida, H.; Sarosi, I.; Tan, H.L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-dos-Santos, A.J.; Van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323.
- Nakagawa, N.; Kinosaki, M.; Yamaguchi, K.; Shima, N.; Yasuda, H.; Yano, K.; Morinaga, T.; Higashio, K. RANK is the essential signaling receptor for osteoclast differentiation factor in osteoclastogenesis. Biochem. Biophys. Res. Commun. 1998, 253, 395–400.
- Schramek, D.; Leibbrandt, A.; Sigl, V.; Kenner, L.; Pospisilik, J.A.; Lee, H.J.; Hanada, R.; Joshi, P.A.; Aliprantis, A.; Glimcher, L.; et al. Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature 2010, 468, 98–102.
- Tanos, T.; Sflomos, G.; Echeverria, P.C.; Ayyanan, A.; Gutierrez, M.; Delaloye, J.F.; Raffoul, W.; Fiche, M.; Dougall, W.; Schneider, P.; et al. Progesterone/RANKL is a major regulatory axis in the human breast. Sci. Transl. Med. 2013, 5, 182ra155.
- Gonzalez-Suarez, E.; Jacob, A.P.; Jones, J.; Miller, R.; Roudier-Meyer, M.P.; Erwert, R.; Pinkas, J.; Branstetter, D.; Dougall, W.C. RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature 2010, 468, 103–107.
- Afzal, M.Z.; Shirai, K. Immune checkpoint inhibitor (anti-CTLA-4, anti-PD-1) therapy alone versus immune checkpoint inhibitor (anti-CTLA-4, anti-PD-1) therapy in combination with anti-RANKL denosumuab in malignant melanoma: A retrospective analysis at a tertiary care center. Melanoma Res. 2018, 28, 341–347.
- Angela, Y.; Haferkamp, S.; Weishaupt, C.; Ugurel, S.; Becker, J.C.; Oberndorfer, F.; Alar, V.; Satzger, I.; Gutzmer, R. Combination of denosumab and immune checkpoint inhibition: Experience in 29 patients with metastatic melanoma and bone metastases. Cancer Immunol. Immunother. 2019, 68, 1187–1194.
- Gomez-Aleza, C.; Gonzalez-Suarez, E. Inhibition of RANK signaling as a potential immunotherapy in breast cancer. Oncoimmunology 2021, 10, 1923156.
- Wong, B.R.; Rho, J.; Arron, J.; Robinson, E.; Orlinick, J.; Chao, M.; Kalachikov, S.; Cayani, E.; Bartlett, F.S., 3rd; Frankel, W.N.; et al. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J. Biol. Chem. 1997, 272, 25190–25194.
- Wong, B.R.; Josien, R.; Lee, S.Y.; Sauter, B.; Li, H.L.; Steinman, R.M.; Choi, Y. TRANCE (tumor necrosis factor [TNF]-related activation-induced cytokine), a new TNF family member predominantly expressed in T cells, is a dendritic cell-specific survival factor. J. Exp. Med. 1997, 186, 2075–2080.
- Hikita, A.; Yana, I.; Wakeyama, H.; Nakamura, M.; Kadono, Y.; Oshima, Y.; Nakamura, K.; Seiki, M.; Tanaka, S. Negative regulation of osteoclastogenesis by ectodomain shedding of receptor activator of NF-kappaB ligand. J. Biol. Chem. 2006, 281, 36846–36855.
- Suzuki, J.; Ikeda, T.; Kuroyama, H.; Seki, S.; Kasai, M.; Utsuyama, M.; Tatsumi, M.; Uematsu, H.; Hirokawa, K. Regulation of osteoclastogenesis by three human RANKL isoforms expressed in NIH3T3 cells. Biochem. Biophys. Res. Commun. 2004, 314, 1021–1027.
- Simonet, W.S.; Lacey, D.L.; Dunstan, C.R.; Kelley, M.; Chang, M.S.; Luthy, R.; Nguyen, H.Q.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell 1997, 89, 309–319.
- Yasuda, H.; Shima, N.; Nakagawa, N.; Mochizuki, S.I.; Yano, K.; Fujise, N.; Sato, Y.; Goto, M.; Yamaguchi, K.; Kuriyama, M.; et al. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): A mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology 1998, 139, 1329–1337.
- Capparelli, C.; Morony, S.; Warmington, K.; Adamu, S.; Lacey, D.; Dunstan, C.R.; Stouch, B.; Martin, S.; Kostenuik, P.J. Sustained antiresorptive effects after a single treatment with human recombinant osteoprotegerin (OPG): A pharmacodynamic and pharmacokinetic analysis in rats. J. Bone Miner. Res. 2003, 18, 852–858.
- Lipton, A.; Goessl, C. Clinical development of anti-RANKL therapies for treatment and prevention of bone metastasis. Bone 2011, 48, 96–99.
- Lacey, D.L.; Boyle, W.J.; Simonet, W.S.; Kostenuik, P.J.; Dougall, W.C.; Sullivan, J.K.; San Martin, J.; Dansey, R. Bench to bedside: Elucidation of the OPG-RANK-RANKL pathway and the development of denosumab. Nat. Rev. Drug Discov. 2012, 11, 401–419.
- Coleman, R.E.; Croucher, P.I.; Padhani, A.R.; Clezardin, P.; Chow, E.; Fallon, M.; Guise, T.; Colangeli, S.; Capanna, R.; Costa, L. Bone metastases. Nat. Rev. Dis. Primers 2020, 6, 83.
- Coleman, R.E. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin. Cancer Res. 2006, 12, 6243s–6249s.
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342.
- Mundy, G.R. Mechanisms of bone metastasis. Cancer 1997, 80, 1546–1556.
- Guise, T.A.; Mohammad, K.S.; Clines, G.; Stebbins, E.G.; Wong, D.H.; Higgins, L.S.; Vessella, R.; Corey, E.; Padalecki, S.; Suva, L.; et al. Basic mechanisms responsible for osteolytic and osteoblastic bone metastases. Clin. Cancer Res. 2006, 12, 6213s–6216s.
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425.
- Yin, J.J.; Mohammad, K.S.; Kakonen, S.M.; Harris, S.; Wu-Wong, J.R.; Wessale, J.L.; Padley, R.J.; Garrett, I.R.; Chirgwin, J.M.; Guise, T.A. A causal role for endothelin-1 in the pathogenesis of osteoblastic bone metastases. Proc. Natl. Acad. Sci. USA 2003, 100, 10954–10959.
- de Groot, A.F.; Appelman-Dijkstra, N.M.; van der Burg, S.H.; Kroep, J.R. The anti-tumor effect of RANKL inhibition in malignant solid tumors—A systematic review. Cancer Treat. Rev. 2018, 62, 18–28.
- Lipton, A.; Fizazi, K.; Stopeck, A.T.; Henry, D.H.; Brown, J.E.; Yardley, D.A.; Richardson, G.E.; Siena, S.; Maroto, P.; Clemens, M.; et al. Superiority of denosumab to zoledronic acid for prevention of skeletal-related events: A combined analysis of 3 pivotal, randomised, phase 3 trials. Eur. J. Cancer 2012, 48, 3082–3092.
- Burgess, T.L.; Qian, Y.; Kaufman, S.; Ring, B.D.; Van, G.; Capparelli, C.; Kelley, M.; Hsu, H.; Boyle, W.J.; Dunstan, C.R.; et al. The ligand for osteoprotegerin (OPGL) directly activates mature osteoclasts. J. Cell Biol. 1999, 145, 527–538.
- Lacey, D.L.; Tan, H.L.; Lu, J.; Kaufman, S.; Van, G.; Qiu, W.; Rattan, A.; Scully, S.; Fletcher, F.; Juan, T.; et al. Osteoprotegerin ligand modulates murine osteoclast survival in vitro and in vivo. Am. J. Pathol. 2000, 157, 435–448.
- Fleisch, H. Development of bisphosphonates. Breast Cancer Res. 2002, 4, 30–34.
- Stopeck, A.T.; Lipton, A.; Body, J.J.; Steger, G.G.; Tonkin, K.; de Boer, R.H.; Lichinitser, M.; Fujiwara, Y.; Yardley, D.A.; Viniegra, M.; et al. Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: A randomized, double-blind study. J. Clin. Oncol. 2010, 28, 5132–5139.
- Fizazi, K.; Carducci, M.; Smith, M.; Damiao, R.; Brown, J.; Karsh, L.; Milecki, P.; Shore, N.; Rader, M.; Wang, H.; et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: A randomised, double-blind study. Lancet 2011, 377, 813–822.
- Henry, D.H.; Costa, L.; Goldwasser, F.; Hirsh, V.; Hungria, V.; Prausova, J.; Scagliotti, G.V.; Sleeboom, H.; Spencer, A.; Vadhan-Raj, S. Randomized, double-blind study of denosumab versus zoledronic acid in the treatment of bone metastases in patients with advanced cancer (excluding breast and prostate cancer) or multiple myeloma. J. Clin. Oncol. 2011, 29, 1125–1132.
- Fournier, P.G.; Stresing, V.; Ebetino, F.H.; Clezardin, P. How do bisphosphonates inhibit bone metastasis in vivo? Neoplasia 2010, 12, 571–578.
- Stresing, V.; Daubine, F.; Benzaid, I.; Monkkonen, H.; Clezardin, P. Bisphosphonates in cancer therapy. Cancer Lett. 2007, 257, 16–35.
- Jones, D.H.; Nakashima, T.; Sanchez, O.H.; Kozieradzki, I.; Komarova, S.V.; Sarosi, I.; Morony, S.; Rubin, E.; Sarao, R.; Hojilla, C.V.; et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature 2006, 440, 692–696.
- Armstrong, A.P.; Miller, R.E.; Jones, J.C.; Zhang, J.; Keller, E.T.; Dougall, W.C. RANKL acts directly on RANK-expressing prostate tumor cells and mediates migration and expression of tumor metastasis genes. Prostate 2008, 68, 92–104.
- Casimiro, S.; Mohammad, K.S.; Pires, R.; Tato-Costa, J.; Alho, I.; Teixeira, R.; Carvalho, A.; Ribeiro, S.; Lipton, A.; Guise, T.A.; et al. RANKL/RANK/MMP-1 molecular triad contributes to the metastatic phenotype of breast and prostate cancer cells in vitro. PLoS ONE 2013, 8, e63153.
- Mori, K.; Le Goff, B.; Charrier, C.; Battaglia, S.; Heymann, D.; Redini, F. DU145 human prostate cancer cells express functional receptor activator of NFkappaB: New insights in the prostate cancer bone metastasis process. Bone 2007, 40, 981–990.
- Mori, K.; Le Goff, B.; Berreur, M.; Riet, A.; Moreau, A.; Blanchard, F.; Chevalier, C.; Guisle-Marsollier, I.; Leger, J.; Guicheux, J.; et al. Human osteosarcoma cells express functional receptor activator of nuclear factor-kappa B. J. Pathol. 2007, 211, 555–562.
- Zheng, Y.; Zhou, H.; Brennan, K.; Blair, J.M.; Modzelewski, J.R.; Seibel, M.J.; Dunstan, C.R. Inhibition of bone resorption, rather than direct cytotoxicity, mediates the anti-tumour actions of ibandronate and osteoprotegerin in a murine model of breast cancer bone metastasis. Bone 2007, 40, 471–478.
- Blake, M.L.; Tometsko, M.; Miller, R.; Jones, J.C.; Dougall, W.C. RANK expression on breast cancer cells promotes skeletal metastasis. Clin. Exp. Metastasis 2014, 31, 233–245.
- Lu, X.; Wang, Q.; Hu, G.; Van Poznak, C.; Fleisher, M.; Reiss, M.; Massague, J.; Kang, Y. ADAMTS1 and MMP1 proteolytically engage EGF-like ligands in an osteolytic signaling cascade for bone metastasis. Genes Dev. 2009, 23, 1882–1894.
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordon-Cardo, C.; Guise, T.A.; Massague, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549.
- Canon, J.; Bryant, R.; Roudier, M.; Branstetter, D.G.; Dougall, W.C. RANKL inhibition combined with tamoxifen treatment increases anti-tumor efficacy and prevents tumor-induced bone destruction in an estrogen receptor-positive breast cancer bone metastasis model. Breast Cancer Res. Treat. 2012, 135, 771–780.
- Gomes, I.; de Almeida, B.P.; Damaso, S.; Mansinho, A.; Correia, I.; Henriques, S.; Cruz-Duarte, R.; Vilhais, G.; Felix, P.; Alves, P.; et al. Expression of receptor activator of NFkB (RANK) drives stemness and resistance to therapy in ER+HER2- breast cancer. Oncotarget 2020, 11, 1714–1728.
- Zhang, J.; Dai, J.; Qi, Y.; Lin, D.L.; Smith, P.; Strayhorn, C.; Mizokami, A.; Fu, Z.; Westman, J.; Keller, E.T. Osteoprotegerin inhibits prostate cancer-induced osteoclastogenesis and prevents prostate tumor growth in the bone. J. Clin. Investig. 2001, 107, 1235–1244.
- Yonou, H.; Kanomata, N.; Goya, M.; Kamijo, T.; Yokose, T.; Hasebe, T.; Nagai, K.; Hatano, T.; Ogawa, Y.; Ochiai, A. Osteoprotegerin/osteoclastogenesis inhibitory factor decreases human prostate cancer burden in human adult bone implanted into nonobese diabetic/severe combined immunodeficient mice. Cancer Res. 2003, 63, 2096–2102.
- Zhang, J.; Dai, J.; Yao, Z.; Lu, Y.; Dougall, W.; Keller, E.T. Soluble receptor activator of nuclear factor kappaB Fc diminishes prostate cancer progression in bone. Cancer Res. 2003, 63, 7883–7890.
- Kiefer, J.A.; Vessella, R.L.; Quinn, J.E.; Odman, A.M.; Zhang, J.; Keller, E.T.; Kostenuik, P.J.; Dunstan, C.R.; Corey, E. The effect of osteoprotegerin administration on the intra-tibial growth of the osteoblastic LuCaP 23.1 prostate cancer xenograft. Clin. Exp. Metastasis 2004, 21, 381–387.
- Asano, T.; Okamoto, K.; Nakai, Y.; Tsutsumi, M.; Muro, R.; Suematsu, A.; Hashimoto, K.; Okamura, T.; Ehata, S.; Nitta, T.; et al. Soluble RANKL is physiologically dispensable but accelerates tumour metastasis to bone. Nat. Metab. 2019, 1, 868–875.
- Pantano, F.; Rossi, E.; Iuliani, M.; Facchinetti, A.; Simonetti, S.; Ribelli, G.; Zoccoli, A.; Vincenzi, B.; Tonini, G.; Zamarchi, R.; et al. Dynamic changes of Receptor activator of nuclear factor-kappaB expression in Circulating Tumor Cells during Denosumab predict treatment effectiveness in Metastatic Breast Cancer. Sci. Rep. 2020, 10, 1288.
- Min, J.K.; Cho, Y.L.; Choi, J.H.; Kim, Y.; Kim, J.H.; Yu, Y.S.; Rho, J.; Mochizuki, N.; Kim, Y.M.; Oh, G.T.; et al. Receptor activator of nuclear factor (NF)-kappaB ligand (RANKL) increases vascular permeability: Impaired permeability and angiogenesis in eNOS-deficient mice. Blood 2007, 109, 1495–1502.
- Wang, J.; Loberg, R.; Taichman, R.S. The pivotal role of CXCL12 (SDF-1)/CXCR4 axis in bone metastasis. Cancer Metastasis Rev. 2006, 25, 573–587.
- Casimiro, S.; Ferreira, A.R.; Mansinho, A.; Alho, I.; Costa, L. Molecular Mechanisms of Bone Metastasis: Which Targets Came from the Bench to the Bedside? Int. J. Mol. Sci. 2016, 17, 1415.
- Liu, F.; Ke, J.; Song, Y. Application of Biomarkers for the Prediction and Diagnosis of Bone Metastasis in Breast Cancer. J. Breast Cancer 2020, 23, 588–598.
- Pavlovic, M.; Arnal-Estape, A.; Rojo, F.; Bellmunt, A.; Tarragona, M.; Guiu, M.; Planet, E.; Garcia-Albeniz, X.; Morales, M.; Urosevic, J.; et al. Enhanced MAF Oncogene Expression and Breast Cancer Bone Metastasis. J. Natl. Cancer Inst. 2015, 107, djv256.
- Westbrook, J.A.; Cairns, D.A.; Peng, J.; Speirs, V.; Hanby, A.M.; Holen, I.; Wood, S.L.; Ottewell, P.D.; Marshall, H.; Banks, R.E.; et al. CAPG and GIPC1: Breast Cancer Biomarkers for Bone Metastasis Development and Treatment. J. Natl. Cancer Inst. 2016, 108.
- Itoh, T.; Ito, Y.; Ohtsuki, Y.; Ando, M.; Tsukamasa, Y.; Yamada, N.; Naoe, T.; Akao, Y. Microvesicles released from hormone-refractory prostate cancer cells facilitate mouse pre-osteoblast differentiation. J. Mol. Histol. 2012, 43, 509–515.
- Canon, J.R.; Roudier, M.; Bryant, R.; Morony, S.; Stolina, M.; Kostenuik, P.J.; Dougall, W.C. Inhibition of RANKL blocks skeletal tumor progression and improves survival in a mouse model of breast cancer bone metastasis. Clin. Exp. Metastasis 2008, 25, 119–129.
- George, C.N.; Canuas-Landero, V.; Theodoulou, E.; Muthana, M.; Wilson, C.; Ottewell, P. Oestrogen and zoledronic acid driven changes to the bone and immune environments: Potential mechanisms underlying the differential anti-tumour effects of zoledronic acid in pre- and post-menopausal conditions. J. Bone Oncol. 2020, 25, 100317.
- Braun, S.; Vogl, F.D.; Naume, B.; Janni, W.; Osborne, M.P.; Coombes, R.C.; Schlimok, G.; Diel, I.J.; Gerber, B.; Gebauer, G.; et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N. Engl. J. Med. 2005, 353, 793–802.
- Cheng, J.N.; Frye, J.B.; Whitman, S.A.; Kunihiro, A.G.; Pandey, R.; Funk, J.L. A Role for TGFbeta Signaling in Preclinical Osteolytic Estrogen Receptor-Positive Breast Cancer Bone Metastases Progression. Int. J. Mol. Sci. 2021, 22, 4463.
- Winding, B.; Misander, H.; Hoegh-Andersen, P.; Brunner, N.; Foged, N.T. Estradiol enhances osteolytic lesions in mice inoculated with human estrogen receptor-negative MDA-231 breast cancer cells in vivo. Breast Cancer Res. Treat. 2003, 78, 205–216.
- Coleman, R.; Cameron, D.; Dodwell, D.; Bell, R.; Wilson, C.; Rathbone, E.; Keane, M.; Gil, M.; Burkinshaw, R.; Grieve, R.; et al. Adjuvant zoledronic acid in patients with early breast cancer: Final efficacy analysis of the AZURE (BIG 01/04) randomised open-label phase 3 trial. Lancet Oncol. 2014, 15, 997–1006.
- Ottewell, P.D.; Wang, N.; Brown, H.K.; Reeves, K.J.; Fowles, C.A.; Croucher, P.I.; Eaton, C.L.; Holen, I. Zoledronic acid has differential antitumor activity in the pre- and postmenopausal bone microenvironment in vivo. Clin. Cancer Res. 2014, 20, 2922–2932.
- Gnant, M.; Pfeiler, G.; Steger, G.G.; Egle, D.; Greil, R.; Fitzal, F.; Wette, V.; Balic, M.; Haslbauer, F.; Melbinger-Zeinitzer, E.; et al. Adjuvant denosumab in postmenopausal patients with hormone receptor-positive breast cancer (ABCSG-18): Disease-free survival results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 339–351.
- Raisz, L.G. Pathogenesis of osteoporosis: Concepts, conflicts, and prospects. J. Clin. Investig. 2005, 115, 3318–3325.
- Hofbauer, L.C.; Rachner, T.D.; Coleman, R.E.; Jakob, F. Endocrine aspects of bone metastases. Lancet Diabetes Endocrinol. 2014, 2, 500–512.
- Melani, C.; Sangaletti, S.; Barazzetta, F.M.; Werb, Z.; Colombo, M.P. Amino-biphosphonate-mediated MMP-9 inhibition breaks the tumor-bone marrow axis responsible for myeloid-derived suppressor cell expansion and macrophage infiltration in tumor stroma. Cancer Res. 2007, 67, 11438–11446.
- Coscia, M.; Quaglino, E.; Iezzi, M.; Curcio, C.; Pantaleoni, F.; Riganti, C.; Holen, I.; Monkkonen, H.; Boccadoro, M.; Forni, G.; et al. Zoledronic acid repolarizes tumour-associated macrophages and inhibits mammary carcinogenesis by targeting the mevalonate pathway. J. Cell Mol. Med. 2010, 14, 2803–2815.
- Benzaid, I.; Monkkonen, H.; Stresing, V.; Bonnelye, E.; Green, J.; Monkkonen, J.; Touraine, J.L.; Clezardin, P. High phosphoantigen levels in bisphosphonate-treated human breast tumors promote Vgamma9Vdelta2 T-cell chemotaxis and cytotoxicity in vivo. Cancer Res. 2011, 71, 4562–4572.
- Benzaid, I.; Monkkonen, H.; Bonnelye, E.; Monkkonen, J.; Clezardin, P. In vivo phosphoantigen levels in bisphosphonate-treated human breast tumors trigger Vgamma9Vdelta2 T-cell antitumor cytotoxicity through ICAM-1 engagement. Clin. Cancer Res. 2012, 18, 6249–6259.
- D’Oronzo, S.; Gregory, W.; Nicholson, S.; Chong, Y.K.; Brown, J.; Coleman, R. Natural history of stage II/III breast cancer, bone metastasis and the impact of adjuvant zoledronate on distribution of recurrences. J. Bone Oncol. 2021, 28, 100367.
- Ferrari-Lacraz, S.; Ferrari, S. Do RANKL inhibitors (denosumab) affect inflammation and immunity? Osteoporos. Int. 2011, 22, 435–446.
- Bucay, N.; Sarosi, I.; Dunstan, C.R.; Morony, S.; Tarpley, J.; Capparelli, C.; Scully, S.; Tan, H.L.; Xu, W.; Lacey, D.L.; et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12, 1260–1268.
- Tomoyasu, A.; Goto, M.; Fujise, N.; Mochizuki, S.; Yasuda, H.; Morinaga, T.; Tsuda, E.; Higashio, K. Characterization of monomeric and homodimeric forms of osteoclastogenesis inhibitory factor. Biochem. Biophys. Res. Commun. 1998, 245, 382–387.
- von Moos, R.; Costa, L.; Gonzalez-Suarez, E.; Terpos, E.; Niepel, D.; Body, J.J. Management of bone health in solid tumours: From bisphosphonates to a monoclonal antibody. Cancer Treat. Rev. 2019, 76, 57–67.
- Coleman, R.; Hadji, P.; Body, J.J.; Santini, D.; Chow, E.; Terpos, E.; Oudard, S.; Bruland, O.; Flamen, P.; Kurth, A.; et al. Bone health in cancer: ESMO Clinical Practice Guidelines. Ann. Oncol. 2020, 31, 1650–1663.
- Mhaskar, R.; Kumar, A.; Miladinovic, B.; Djulbegovic, B. Bisphosphonates in multiple myeloma: An updated network meta-analysis. Cochrane Database Syst. Rev. 2017, 12, CD003188.
- Smith, M.R.; Halabi, S.; Ryan, C.J.; Hussain, A.; Vogelzang, N.; Stadler, W.; Hauke, R.J.; Monk, J.P.; Saylor, P.; Bhoopalam, N.; et al. Randomized controlled trial of early zoledronic acid in men with castration-sensitive prostate cancer and bone metastases: Results of CALGB 90202 (alliance). J. Clin. Oncol. 2014, 32, 1143–1150.
- Raje, N.; Terpos, E.; Willenbacher, W.; Shimizu, K.; Garcia-Sanz, R.; Durie, B.; Legiec, W.; Krejci, M.; Laribi, K.; Zhu, L.; et al. Denosumab versus zoledronic acid in bone disease treatment of newly diagnosed multiple myeloma: An international, double-blind, double-dummy, randomised, controlled, phase 3 study. Lancet Oncol. 2018, 19, 370–381.
- Henry, D.; Vadhan-Raj, S.; Hirsh, V.; von Moos, R.; Hungria, V.; Costa, L.; Woll, P.J.; Scagliotti, G.; Smith, G.; Feng, A.; et al. Delaying skeletal-related events in a randomized phase 3 study of denosumab versus zoledronic acid in patients with advanced cancer: An analysis of data from patients with solid tumors. Support. Care Cancer 2014, 22, 679–687.
- Peters, S.; Danson, S.; Hasan, B.; Dafni, U.; Reinmuth, N.; Majem, M.; Tournoy, K.G.; Mark, M.T.; Pless, M.; Cobo, M.; et al. A Randomized Open-Label Phase III Trial Evaluating the Addition of Denosumab to Standard First-Line Treatment in Advanced NSCLC: The European Thoracic Oncology Platform (ETOP) and European Organisation for Research and Treatment of Cancer (EORTC) SPLENDOUR Trial. J. Thorac. Oncol. 2020, 15, 1647–1656.
- Scagliotti, G.V.; Hirsh, V.; Siena, S.; Henry, D.H.; Woll, P.J.; Manegold, C.; Solal-Celigny, P.; Rodriguez, G.; Krzakowski, M.; Mehta, N.D.; et al. Overall survival improvement in patients with lung cancer and bone metastases treated with denosumab versus zoledronic acid: Subgroup analysis from a randomized phase 3 study. J. Thorac. Oncol. 2012, 7, 1823–1829.
- Morgan, G.J.; Davies, F.E.; Gregory, W.M.; Bell, S.E.; Szubert, A.J.; Cook, G.; Drayson, M.T.; Owen, R.G.; Ross, F.M.; Jackson, G.H.; et al. Long-term follow-up of MRC Myeloma IX trial: Survival outcomes with bisphosphonate and thalidomide treatment. Clin. Cancer Res. 2013, 19, 6030–6038.
- Raje, N.; Vadhan-Raj, S.; Willenbacher, W.; Terpos, E.; Hungria, V.; Spencer, A.; Alexeeva, Y.; Facon, T.; Stewart, A.K.; Feng, A.; et al. Evaluating results from the multiple myeloma patient subset treated with denosumab or zoledronic acid in a randomized phase 3 trial. Blood Cancer J. 2016, 6, e378.
- Tsourdi, E.; Langdahl, B.; Cohen-Solal, M.; Aubry-Rozier, B.; Eriksen, E.F.; Guanabens, N.; Obermayer-Pietsch, B.; Ralston, S.H.; Eastell, R.; Zillikens, M.C. Discontinuation of Denosumab therapy for osteoporosis: A systematic review and position statement by ECTS. Bone 2017, 105, 11–17.
- Cummings, S.R.; Ferrari, S.; Eastell, R.; Gilchrist, N.; Jensen, J.B.; McClung, M.; Roux, C.; Torring, O.; Valter, I.; Wang, A.T.; et al. Vertebral Fractures After Discontinuation of Denosumab: A Post Hoc Analysis of the Randomized Placebo-Controlled FREEDOM Trial and Its Extension. J. Bone Miner. Res. 2018, 33, 190–198.
More
Information
Subjects:
Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
825
Revisions:
2 times
(View History)
Update Date:
10 Aug 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No