 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Christina Nedev | + 4547 word(s) | 4547 | 2021-07-12 08:24:43 | | | |
| 2 | Nora Tang | Meta information modification | 4547 | 2021-08-05 08:06:17 | | |
Video Upload Options
Sepsis is a life-threatening medical condition that occurs when the host has an uncontrolled or abnormal immune response to overwhelming infection. It is now widely accepted that sepsis occurs in two concurrent phases, which consist of an initial immune activation phase followed by a chronic immunosuppressive phase, leading to immune cell death. Depending on the severity of the disease and the pathogen involved, the hosts immune system may not fully recover, leading to ongoing complications proceeding the initial infection. As such, sepsis remains one of the leading causes of morbidity and mortality world-wide, with treatment options limited to general treatment in intensive care units (ICU). Lack of specific treatments available for sepsis is mostly due to our limited knowledge of the immuno-physiology associated with the disease. This review will provide a comprehensive overview of the mechanisms and cell types involved in eliciting infection-induced immune activation from both the innate and adaptive immune system during sepsis. In addition, the mechanisms leading to immune cell death following hyperactivation of immune cells will be explored. The evaluation and better understanding of the cellular and systemic responses leading to disease onset could eventuate into the development of much needed therapies to combat this unrelenting disease.
1. Introduction
2. Immune Activation and Sepsis
2.1. Acute Innate Immune Response
2.2. Acute Adaptive Immune Response
3. Immune Suppression and Cell Death during Sepsis
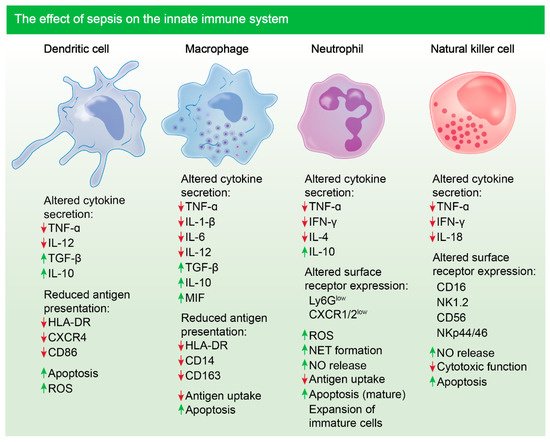
3.1. Chronic Innate Immune Response
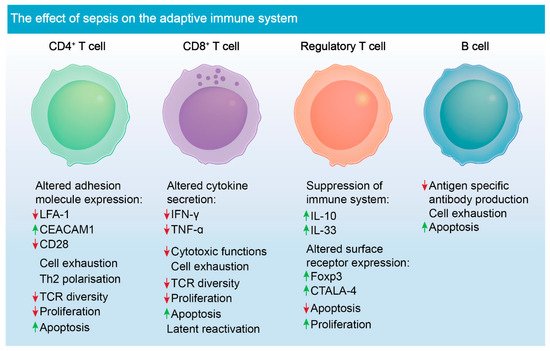
3.2. Chronic Adaptive Immune Response
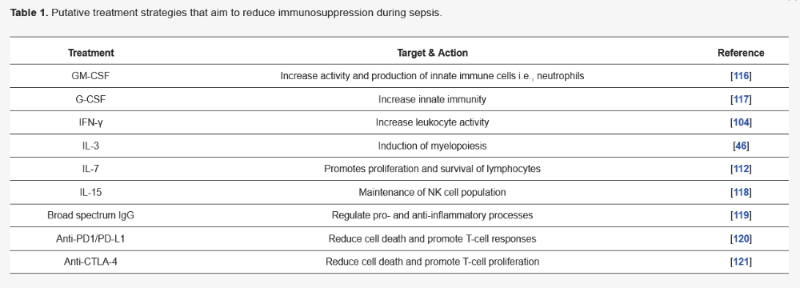
4. Putative Therapies and Biomarkers Targeting Immune Suppression during Sepsis
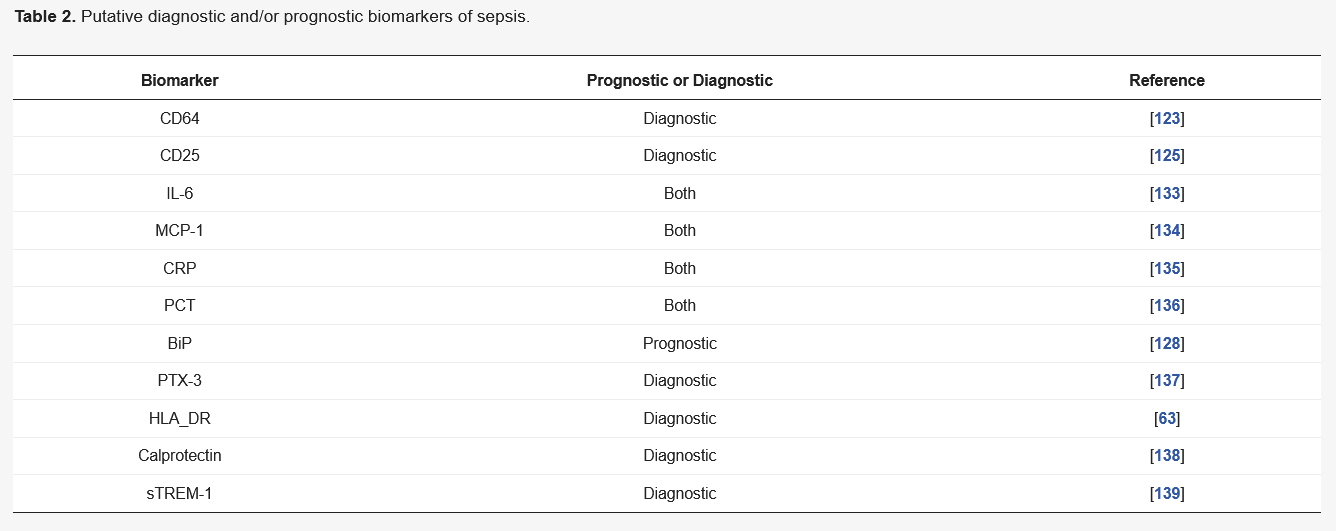
5. Conclusions
References
- Gotts, J.E.; Matthay, M.A. Sepsis: Pathophysiology and clinical management. BMJ 2016, 353, i1585.
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211.
- Venet, F.; Monneret, G. Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat. Rev. Nephrol. 2018, 14, 121–137.
- Nedeva, C.; Menassa, J.; Puthalakath, H. Sepsis: Inflammation Is a Necessary Evil. Front. Cell Dev. Biol. 2019, 7, 108.
- Delano, M.J.; Ward, P.A. The immune system’s role in sepsis progression, resolution, and long-term outcome. Immunol. Rev. 2016, 274, 330–353.
- Boomer, J.S.; Green, J.M.; Hotchkiss, R.S. The changing immune system in sepsis: Is individualized immuno-modulatory therapy the answer? Virulence 2014, 5, 45–56.
- Nedeva, C.; Menassa, J.; Duan, M.; Liu, C.; Doerflinger, M.; Kueh, A.J.; Herold, M.J.; Fonseka, P.; Phan, T.K.; Faou, P.; et al. TREML4 receptor regulates inflammation and innate immune cell death during polymicrobial sepsis. Nat. Immunol. 2020, 21, 1585–1596.
- Doerflinger, M.; Glab, J.; Nedeva, C.; Jose, I.; Lin, A.; O’Reilly, L.; Allison, C.; Pellegrini, M.; Hotchkiss, R.S.; Puthalakath, H. Chemical chaperone TUDCA prevents apoptosis and improves survival during polymicrobial sepsis in mice. Sci. Rep. 2016, 6, 34702.
- Rentz, A.M.; Halpern, M.; Bowden, R. The Impact of Candidemia on Length of Hospital Stay, Outcome, and Overall Cost of Illness. Clin. Infect. Dis. 1998, 27, 781–788.
- Olwal, C.O.; Nganyewo, N.N.; Tapela, K.; Zune, A.L.D.; Owoicho, O.; Bediako, Y.; Duodu, S. Parallels in Sepsis and COVID-19 Conditions: Implications for Managing Severe COVID-19. Front. Immunol. 2021, 12, 602848.
- Zhao, G.-J.; Li, D.; Zhao, Q.; Song, J.-X.; Chen, X.-R.; Hong, G.-L.; Li, M.-F.; Wu, B.; Lu, Z. Incidence, risk factors and impact on outcomes of secondary infection in patients with septic shock: An 8-year retrospective study. Sci. Rep. 2016, 6, 38361.
- Lin, G.-L.; McGinley, J.P.; Drysdale, S.B.; Pollard, A.J. Epidemiology and Immune Pathogenesis of Viral Sepsis. Front. Immunol. 2018, 9, 2147.
- Horan, T.C.; Andrus, M.; Dudeck, M.A. CDC/NHSN surveillance definition of health care–associated infection and criteria for specific types of infections in the acute care setting. Am. J. Infect. Control. 2008, 36, 309–332.
- Otto, G.P.; Sossdorf, M.; Claus, R.A.; Rödel, J.; Menge, K.; Reinhart, K.; Bauer, M.; Riedemann, N.C. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit. Care 2011, 15, R183.
- Daviaud, F.; Grimaldi, D.; Dechartres, A.; Charpentier, J.; Geri, G.; Marin, N.; Chiche, J.-D.; Cariou, A.; Mira, J.-P.; Pène, F. Timing and causes of death in septic shock. Ann. Intensiv. Care 2015, 5, 16.
- Arens, C.; Bajwa, S.A.; Koch, C.; Siegler, B.H.; Schneck, E.; Hecker, A.; Weiterer, S.; Lichtenstern, C.; Weigand, M.A.; Uhle, F. Sepsis-induced long-term immune paralysis–results of a descriptive, explorative study. Crit. Care 2016, 20, 93.
- Marik, P.E.; Linde-Zwirble, W.T.; Bittner, E.A.; Sahatjian, J.; Hansell, D. Fluid administration in severe sepsis and septic shock, patterns and outcomes: An analysis of a large national database. Intensiv. Care Med. 2017, 43, 625–632.
- Rhodes, A.A.; Evans, L.E.; Alhazzani, W.; Levy, M.M.; Antonelli, M.; Ferrer, R.; Kumar, A.; Sevransky, J.E.; Sprung, C.L.; Nunnally, M.E.; et al. Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock: 2016. Crit. Care Med. 2017, 45, 486–552.
- Marshall, J.C. Special issue: Sepsis Why have clinical trials in sepsis failed? Trends Mol. Med. 2014, 20, 195–203.
- Menassa, J.; Nedeva, C.; Pollock, C.; Puthalakath, H. TLR4: The fall guy in sepsis? Cell Stress 2020, 4, 270–272.
- Thomas, L. Germs. N. Engl. J. Med. 1972, 287, 553–555.
- Akira, S. Pathogen recognition by innate immunity and its signaling. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 143–156.
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987–995.
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. 1986. J. Immunol. 2005, 175, 5–14.
- Hailman, E.; Lichenstein, H.S.; Wurfel, M.M.; Miller, D.S.; Johnson, D.A.; Kelley, M.; Busse, L.A.; Zukowski, M.M.; Wright, S.D. Lipopolysaccharide (LPS)-binding protein accelerates the binding of LPS to CD14. J. Exp. Med. 1994, 179, 269–277.
- Schromm, A.B.; Lien, E.; Henneke, P.; Chow, J.C.; Yoshimura, A.; Heine, H.; Latz, E.; Monks, B.G.; Schwartz, D.A.; Miyake, K.; et al. Molecular genetic analysis of an endotoxin nonresponder mutant cell line: A point mutation in a conserved region of MD-2 abolishes endotoxin-induced signaling. J. Exp. Med. 2001, 194, 79–88.
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008.
- Sabroe, I.; Williams, T.J.; Hébert, C.A.; Collins, P.D.; Sabroe, I.; Williams, T.J.; Hébert, C.A.; Collins, P.D. Chemoattractant cross-desensitization of the human neutrophil IL-8 receptor involves receptor internalization and differential receptor subtype regulation. J. Immunol. 1997, 158, 1361–1369.
- Nicholson, M.W.; Barclay, A.N.; Singer, M.S.; Rosen, S.D.; van der Merwe, P.A. Affinity and Kinetic Analysis of L-selectin (CD62L) Binding to Glycosylation-dependent Cell-adhesion Molecule-1. J. Biol. Chem. 1998, 273, 763–770.
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535.
- Mantegazza, A.R.; Magalhaes, J.G.; Amigorena, S.; Marks, M.S. Presentation of phagocytosed antigens by MHC class I and II. Traffic 2013, 14, 135–152.
- Simmons, J.; Pittet, J.-F. The coagulopathy of acute sepsis. Curr. Opin. Anaesthesiol. 2015, 28, 227–236.
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern Recognition Receptors and the Innate Immune Response to Viral Infection. Viruses 2011, 3, 920–940.
- Costa-Pereira, A.P.; Williams, T.M.; Strobl, B.; Watling, D.; Briscoe, J.; Kerr, I.M. The Antiviral Response to Gamma Interferon. J. Virol. 2002, 76, 9060–9068.
- Stambas, J.; Lu, C.; Tripp, R. Innate and adaptive immune responses in respiratory virus infection: Implications for the clinic. Expert Rev. Respir. Med. 2020, 14, 1141–1147.
- Bone, R.C.; Balk, R.A.; Cerra, F.B.; Dellinger, R.P.; Fein, A.M.; Knaus, W.A.; Schein, R.M.; Sibbald, W.J. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Chest 1992, 101, 1644–1655.
- Teijaro, J.R.; Walsh, K.B.; Cahalan, S.; Fremgen, D.M.; Roberts, E.; Scott, F.; Martinborough, E.; Peach, R.; Oldstone, M.B.; Rosen, H. Endothelial Cells Are Central Orchestrators of Cytokine Amplification during Influenza Virus Infection. Cell 2011, 146, 980–991.
- Tissari, J.; Sirén, J.; Meri, S.; Julkunen, I.; Matikainen, S. IFN-α Enhances TLR3-Mediated Antiviral Cytokine Expression in Human Endothelial and Epithelial Cells by Up-Regulating TLR3 Expression. J. Immunol. 2005, 174, 4289–4294.
- Andonegui, G.; Bonder, C.S.; Green, F.; Mullaly, S.C.; Zbytnuik, L.; Raharjo, E.; Kubes, P. Endothelium-derived Toll-like receptor-4 is the key molecule in LPS-induced neutrophil sequestration into lungs. J. Clin. Investig. 2003, 111, 1011–1020.
- Karakike, E.; Giamarellos-Bourboulis, E.J. Macrophage Activation-Like Syndrome: A Distinct Entity Leading to Early Death in Sepsis. Front. Immunol. 2019, 10, 55.
- Salvemini, D.; Cuzzocrea, S. Oxidative stress in septic shock and disseminated intravascular coagulation. Free Radic Biol. Med. 2002, 33, 1173–1185.
- Ward, P.A. The Harmful Role of C5a on Innate Immunity in Sepsis. J. Innate Immun. 2010, 2, 439–445.
- Martinez-Sanchez, M.E.; Huerta, L.; Alvarez-Buylla, E.R.; Luján, C.V. Role of Cytokine Combinations on CD4+ T Cell Differentiation, Partial Polarization, and Plasticity: Continuous Network Modeling Approach. Front. Physiol. 2018, 9, 877.
- Tham, E.L.; Shrikant, P.; Mescher, M.F. Activation-Induced Nonresponsiveness: A Th-Dependent Regulatory Checkpoint in the CTL Response. J. Immunol. 2002, 168, 1190–1197.
- Xue, M.; Xie, J.; Liu, L.; Huang, Y.; Guo, F.; Xu, J.; Yang, Y.; Qiu, H. Early and dynamic alterations of Th2/Th1 in previously immunocompetent patients with community-acquired severe sepsis: A prospective observational study. J. Transl. Med. 2019, 17, 57.
- Weber, G.F.; Chousterman, B.G.; He, S.; Fenn, A.M.; Nairz, M.; Anzai, A.; Brenner, T.; Uhle, F.; Iwamoto, Y.; Robbins, C.S.; et al. Interleukin-3 amplifies acute inflammation and is a potential therapeutic target in sepsis. Science 2015, 347, 1260–1265.
- Zhang, X.; Gao, L.; Lei, L.; Zhong, Y.; Dube, P.; Berton, M.T.; Arulanandam, B.; Zhang, J.; Zhong, G. A MyD88-Dependent Early IL-17 Production Protects Mice against Airway Infection with the Obligate Intracellular PathogenChlamydia muridarum. J. Immunol. 2009, 183, 1291–1300.
- Jiang, J.; Zenewicz, L.A.; Mateo, L.R.S.; Lau, L.L.; Shen, H. Activation of Antigen-Specific CD8 T Cells Results in Minimal Killing of Bystander Bacteria. J. Immunol. 2003, 171, 6032–6038.
- Maurice, N.J.; McElrath, M.J.; Andersen-Nissen, E.; Frahm, N.; Prlic, M. CXCR3 enables recruitment and site-specific bystander activation of memory CD8+ T cells. Nat. Commun. 2019, 10, 4987.
- Hotchkiss, R.S.; Tinsley, K.W.; Swanson, P.E.; Schmieg, R.E.; Hui, J.J.; Chang, K.C.; Osborne, D.F.; Freeman, B.D.; Cobb, J.P.; Buchman, T.G.; et al. Sepsis-Induced Apoptosis Causes Progressive Profound Depletion of B and CD4+T Lymphocytes in Humans. J. Immunol. 2001, 166, 6952–6963.
- Andreu-Ballester, J.C.; Tormo-Calandín, C.; Garcia-Ballesteros, C.; Pérez-Griera, J.; Amigó, V.; Almela-Quilis, A.; del Castillo, J.R.; Peñarroja-Otero, C.; Ballester, F. Association of gammadelta T cells with disease severity and mortality in septic patients. Clin. Vaccine Immunol. 2013, 20, 738–746.
- Kelly-Scumpia, K.M.; Scumpia, P.O.; Weinstein, J.S.; Delano, M.; Cuenca, A.G.; Nacionales, D.C.; Wynn, J.; Lee, P.Y.; Kumagai, Y.; Efron, P.A.; et al. B cells enhance early innate immune responses during bacterial sepsis. J. Exp. Med. 2011, 208, 1673–1682.
- Macháček, T.; Panská, L.; Dvořáková, H.; Horák, P. Nitric oxide and cytokine production by glial cells exposed in vitro to neuropathogenic schistosome Trichobilharzia regenti. Parasites Vectors 2016, 9, 579.
- Smeekens, S.P.; Ng, A.; Kumar, V.; Johnson, M.; Plantinga, T.; van Diemen, C.; Arts, P.; Verwiel, E.T.P.; Gresnigt, M.; Fransen, K.; et al. Functional genomics identifies type I interferon pathway as central for host defense against Candida albicans. Nat. Commun. 2013, 4, 1342.
- Skrupky, L.P.; Kerby, P.W.; Hotchkiss, R.S. Advances in the Management of Sepsis and the Understanding of Key Immunologic Defects. Surv. Anesthesiol. 2012, 56, 273–275.
- Rosales, C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Front. Physiol. 2018, 9, 113.
- Vono, M.; Lin, A.; Norrby-Teglund, A.; Koup, R.A.; Liang, F.; Loré, K. Neutrophils acquire the capacity for antigen presentation to memory CD4(+) T cells in vitro and ex vivo. Blood 2017, 129, 1991–2001.
- Basu, S.; Hodgson, G.; Katz, M.; Dunn, A.R. Evaluation of role of G-CSF in the production, survival, and release of neutrophils from bone marrow into circulation. Blood 2002, 100, 854–861.
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Cell. Infect. Microbiol. 2017, 7, 373.
- Guo, R.-F.; Sun, L.; Gao, H.; Shi, K.X.; Rittirsch, D.; Sarma, V.J.; Zetoune, F.S.; Ward, P.A. In vivo regulation of neutrophil apoptosis by C5a during sepsis. J. Leukoc. Biol. 2006, 80, 1575–1583.
- Munoz, C.; Carlet, J.; Fitting, C.; Misset, B.; Blériot, J.P.; Cavaillon, J.M.; Munoz, C.; Carlet, J.; Fitting, C.; Misset, B.; et al. Dysregulation of in vitro cytokine production by monocytes during sepsis. J. Clin. Investig. 1991, 88, 1747–1754.
- Kessler, B.; Rinchai, D.; Kewcharoenwong, C.; Nithichanon, A.; Biggart, R.; Hawrylowicz, C.; Bancroft, G.J.; Lertmemongkolchai, G. Interleukin 10 inhibits pro-inflammatory cytokine responses and killing of Burkholderia pseudomallei. Sci. Rep. 2017, 7, srep42791.
- Winkler, M.S.; Rissiek, A.; Priefler, M.; Schwedhelm, E.; Robbe, L.; Bauer, A.; Zahrte, C.; Zoellner, C.; Kluge, S.; Nierhaus, A. Human leucocyte antigen (HLA-DR) gene expression is reduced in sepsis and correlates with impaired TNFalpha response: A diagnostic tool for immunosuppression? PLoS ONE 2017, 12, e0182427.
- Chalifour, A.; Jeannin, P.; Gauchat, J.-F.; Blaecke, A.; Malissard, M.; N’Guyen, T.; Thieblemont, N.; Delneste, Y. Direct bacterial protein PAMP recognition by human NK cells involves TLRs and triggers α-defensin production. Blood 2004, 104, 1778–1783.
- Puente, J.; Carvajal, T.; Parra, S.; Miranda, D.; Sepulveda, C.; Wolf, M.E.; Mosnaim, A.D. In vitro studies of natural killer cell activity in septic shock patients. Response to a challenge with alpha-interferon and interleukin-2. Int. J. Clin. Pharmacol. Ther. Toxicol. 1993, 31, 271–275.
- Holub, M.; Klučková, Z.; Helcl, M.; Příahodov, J.; Rokyta, R.; Beran, O. Lymphocyte subset numbers depend on the bacterial origin of sepsis. Clin. Microbiol. Infect. 2003, 9, 202–211.
- Chiche, L.; Forel, J.; Thomas, G.; Farnarier, C.; Cognet, C.; Guervilly, C.; Zandotti, C.; Vély, F.; Roch, A.; Vivier, E.; et al. Interferon-gamma production by natural killer cells and cytomegalovirus in critically ill patients. Crit. Care Med. 2012, 40, 3162–3169.
- Faivre, V.; Lukaszewicz, A.; Alves, A.; Charron, D.; Payen, D.; Haziot, A. Accelerated in vitro differentiation of blood monocytes into dendritic cells in human sepsis. Clin. Exp. Immunol. 2007, 147, 426–439.
- Strother, R.K.; Danahy, D.B.; Kotov, D.I.; Kucaba, T.A.; Zacharias, Z.R.; Griffith, T.S.; Legge, K.; Badovinac, V.P. Polymicrobial Sepsis Diminishes Dendritic Cell Numbers and Function Directly Contributing to Impaired Primary CD8 T Cell Responses In Vivo. J. Immunol. 2016, 197, 4301–4311.
- Steinbrink, K.; Graulich, E.; Kubsch, S.; Knop, J.; Enk, A.H. CD4+ and CD8+ anergic T cells induced by interleukin-10–treated human dendritic cells display antigen-specific suppressor activity. Blood 2002, 99, 2468–2476.
- Faivre, V.; Lukaszewicz, A.C.; Alvès, A.; Charron, M.; Payen, D.; Haziot, A. Human Monocytes Differentiate into Dendritic Cells Subsets that Induce Anergic and Regulatory T Cells in Sepsis. PLoS ONE 2012, 7, e47209.
- Weber, G.F.; Maier, S.L.; Zönnchen, T.; Breucha, M.; Seidlitz, T.; Kutschick, I.; Weitz, J. Analysis of circulating plasmacytoid dendritic cells during the course of sepsis. Surgery 2015, 158, 248–254.
- Novotny, A.R.; Reim, D.; Assfalg, V.; Altmayr, F.; Friess, H.M.; Emmanuel, K.; Holzmann, B. Mixed antagonist response and sepsis severity-dependent dysbalance of pro- and anti-inflammatory responses at the onset of postoperative sepsis. Immunobiology 2012, 217, 616–621.
- Gautam, A.; Dixit, S.; Embers, M.; Gautam, R.; Philipp, M.T.; Singh, S.R.; Morici, L.; Dennis, V.A. Different Patterns of Expression and of IL-10 Modulation of Inflammatory Mediators from Macrophages of Lyme Disease-Resistant and -Susceptible Mice. PLoS ONE 2012, 7, e43860.
- Chaudhry, H.; Zhou, J.; Zhong, Y.; Ali, M.M.; McGuire, F.; Nagarkatti, P.S.; Nagarkatti, M. Role of cytokines as a double-edged sword in sepsis. In Vivo 2013, 27, 669–684.
- Kapellos, T.S.; Bonaguro, L.; Gemünd, I.; Reusch, N.; Saglam, A.; Hinkley, E.R.; Schultze, J.L. Human Monocyte Subsets and Phenotypes in Major Chronic Inflammatory Diseases. Front. Immunol. 2019, 10, 2035.
- Baudesson de Chanville, C.; Chousterman, B.G.; Hamon, P.; Laviron, M.; Guillou, N.; Loyher, P.L.; Meghraoui-Kheddar, A.; Barthelemy, S.; Deterre, P.; Boissonnas, A.; et al. Sepsis Triggers a Late Expansion of Functionally Impaired Tissue-Vascular Inflammatory Monocytes During Clinical Recovery. Front. Immunol. 2020, 11, 675.
- Byrne, A.; Reen, D.J. Lipopolysaccharide Induces Rapid Production of IL-10 by Monocytes in the Presence of Apoptotic Neutrophils. J. Immunol. 2002, 168, 1968–1977.
- Schulte, W.; Bernhagen, J.; Bucala, R. Cytokines in Sepsis: Potent Immunoregulators and Potential Therapeutic Targets—An Updated View. Mediat. Inflamm. 2013, 2013, 165974.
- Guo, Y.; Patil, N.K.; Luan, L.; Bohannon, J.K.; Sherwood, E.R. The biology of natural killer cells during sepsis. Immunology 2017, 153, 190–202.
- Cao, C.; Yu, M.; Chai, Y. Pathological alteration and therapeutic implications of sepsis-induced immune cell apoptosis. Cell Death Dis. 2019, 10, 782.
- Pepper, M.; Jenkins, M. Origins of CD4+ effector and central memory T cells. Nat. Immunol. 2011, 12, 467–471.
- Schmoeckel, K.; Traffehn, S.; Eger, C.; Pötschke, C.; Bröker, B. Full Activation of CD4+ T Cells Early During Sepsis Requires Specific Antigen. Shock 2015, 43, 192–200.
- Drewry, A.M.; Samra, N.; Skrupky, L.P.; Fuller, B.; Compton, S.M.; Hotchkiss, R.S. Persistent Lymphopenia After Diagnosis of Sepsis Predicts Mortality. Shock 2014, 42, 383–391.
- Hotchkiss, R.S.; Opal, S.M. Immunotherapy for Sepsis—A New Approach against an Ancient Foe. N. Engl. J. Med. 2010, 363, 87–89.
- Unsinger, J.; McGlynn, M.; Kasten, K.R.; Hoekzema, A.S.; Watanabe, E.; Muenzer, J.T.; McDonough, J.S.; Tschoep, J.; Ferguson, T.A.; McDunn, J.; et al. IL-7 Promotes T Cell Viability, Trafficking, and Functionality and Improves Survival in Sepsis. J. Immunol. 2010, 184, 3768–3779.
- Gershon, R.K.; Kondo, K. Cell interactions in the induction of tolerance: The role of thymic lymphocytes. Immunology 1970, 18, 723–737.
- Venet, F.; Pachot, A.; Debard, A.-L.; Bohe, J.; Bienvenu, J.; Lepape, A.; Powell, W.; Monneret, G. Human CD4+CD25+ Regulatory T Lymphocytes Inhibit Lipopolysaccharide-Induced Monocyte Survival through a Fas/Fas Ligand-Dependent Mechanism. J. Immunol. 2006, 177, 6540–6547.
- Mazer, M.; Unsinger, J.; Drewry, A.; Walton, A.; Osborne, D.; Blood, T.; Hotchkiss, R.; Remy, K.E. IL-10 Has Differential Effects on the Innate and Adaptive Immune Systems of Septic Patients. J. Immunol. 2019, 203, 2088–2099.
- Cao, C.; Chai, Y.; Shou, S.; Wang, J.; Huang, Y.; Ma, T. Toll-like receptor 4 deficiency increases resistance in sepsis-induced immune dysfunction. Int. Immunopharmacol. 2018, 54, 169–176.
- Betts, R.J.; Ho, A.W.S.; Kemeny, D.M. Partial Depletion of Natural CD4+CD25+ Regulatory T Cells with Anti-CD25 Antibody Does Not Alter the Course of Acute Influenza A Virus Infection. PLoS ONE 2011, 6, e27849.
- Akkaya, M.; Kwak, K.; Pierce, S.K. B cell memory: Building two walls of protection against pathogens. Nat. Rev. Immunol. 2020, 20, 229–238.
- Rauch, P.J.; Chudnovskiy, A.; Robbins, C.S.; Weber, G.F.; Etzrodt, M.; Hilgendorf, I.; Tiglao, E.; Figueiredo, J.; Iwamoto, Y.; Theurl, I.; et al. Innate response activator B cells protect against microbial sepsis. Science 2012, 335, 597–601.
- Deng, Q.; Zhao, T.; Pan, B.; Dennahy, I.S.; Duan, X.; Williams, A.M.; Liu, B.; Lin, N.; Bhatti, U.F.; Chen, E.; et al. Protective Effect of Tubastatin A in CLP-Induced Lethal Sepsis. Inflammation 2018, 41, 2101–2109.
- Van der Flier, M.; Sharma, D.B.; Estevão, S.; Emonts, M.; Rook, D.; Hazelzet, J.; van Goudoever, J.B.; Hartwig, N.G. Increased CD4(+) T cell co-inhibitory immune receptor CEACAM1 in neonatal sepsis and soluble-CEACAM1 in meningococcal sepsis: A role in sepsis-associated immune suppression? PLoS ONE 2013, 8, e68294.
- Park, S.W.; Royal, W., III; Semba, R.D.; Wiegand, G.W.; Griffin, D.E. Expression of adhesion molecules and CD28 on T lymphocytes during human immunodeficiency virus infection. Clin. Diagn. Lab. Immunol. 1998, 5, 583–587.
- Jensen, I.J.; Sjaastad, F.V.; Griffith, T.S.; Badovinac, V.P. Sepsis-Induced T Cell Immunoparalysis: The Ins and Outs of Impaired T Cell Immunity. J. Immunol. 2018, 200, 1543–1553.
- Jiang, L.N.; Yao, Y.M.; Sheng, Z.Y. The role of regulatory T cells in the pathogenesis of sepsis and its clinical implication. J. Interferon Cytokine Res. 2012, 32, 341–349.
- Nascimento, D.C.; Melo, P.H.; Piñeros, A.R.; Ferreira, R.G.; Colon, D.; Donate, P.B.; Castanheira, F.V.; Gozzi, A.; Czaikoski, P.G.; Niedbala, W.; et al. IL-33 contributes to sepsis-induced long-term immunosuppression by expanding the regulatory T cell population. Nat. Commun. 2017, 8, 14919.
- Steinhagen, F.; Schmidt, S.V.; Schewe, J.-C.; Peukert, K.; Klinman, D.M.; Bode, C. Immunotherapy in sepsis-brake or accelerate? Pharmacol. Ther. 2020, 208, 107476.
- Shibata, Y.; Berclaz, P.-Y.; Chroneos, Z.C.; Yoshida, M.; Whitsett, J.A.; Trapnell, B.C. GM-CSF Regulates Alveolar Macrophage Differentiation and Innate Immunity in the Lung through PU.1. Immunity 2001, 15, 557–567.
- Gennari, R.; Alexander, J.W.; Gianotti, L.; Eaves-Pyles, T.; Hartmann, S. Granulocyte Macrophage Colony-Stimulating Factor Improves Survival in Two Models of Gut-Derived Sepsis by Improving Gut Barrier Function and Modulating Bacterial Clearance. Ann. Surg. 1994, 220, 68–76.
- Flohe, S.B.; Agrawal, H.; Flohé, S.; Rani, M.; Bangen, J.M.; Schade, F.U. Diversity of interferon gamma and granulocyte-macrophage colony-stimulating factor in restoring immune dysfunction of dendritic cells and macrophages during polymicrobial sepsis. Mol. Med. 2008, 14, 247–256.
- Payen, D.; Faivre, V.; Miatello, J.; Leentjens, J.; Brumpt, C.; Tissières, P.; Dupuis, C.; Pickkers, P.; Lukaszewicz, A.C. Multicentric experience with interferon gamma therapy in sepsis induced immunosuppression. A case series. BMC Infect. Dis. 2019, 19, 931.
- Bo, L.; Wang, F.; Zhu, J.; Li, J.; Deng, X. Granulocyte-colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF) for sepsis: A meta-analysis. Crit. Care 2011, 15, R58.
- Carr, R.; Modi, N.; Dore, C. G-CSF and GM-CSF for treating or preventing neonatal infections. Cochrane Database Syst. Rev. 2003, 2003, CD003066.
- Williams, G.T.; Smith, C.A.; Spooncer, E.; Dexter, T.M.; Taylor, D.R. Haemopoietic colony stimulating factors promote cell survival by suppressing apoptosis. Nature 1990, 343, 76–79.
- Weber, G.F.; Chousterman, B.G.; Hilgendorf, I.; Robbins, C.S.; Theurl, I.; Gerhardt, L.M.S.; Iwamoto, Y.; Quach, T.D.; Ali, M.; Chen, J.W.; et al. Pleural innate response activator B cells protect against pneumonia via a GM-CSF-IgM axis. J. Exp. Med. 2014, 211, 1243–1256.
- Bentzer, P.; Fjell, C.; Walley, K.R.; Boyd, J.; Russell, J.A. Plasma cytokine levels predict response to corticosteroids in septic shock. Intensiv. Care Med. 2016, 42, 1970–1979.
- Cuenca, A.G.; Cuenca, A.L.; Gentile, L.F.; Efron, P.A.; Islam, S.; Moldawer, L.L.; Kays, D.W.; Larson, S.D. Delayed emergency myelopoiesis following polymicrobial sepsis in neonates. Innate Immun. 2015, 21, 386–391.
- Shindo, Y.; Fuchs, A.G.; Davis, C.G.; Eitas, T.; Unsinger, J.; Burnham, C.-A.D.; Green, J.M.; Morre, M.; Bochicchio, G.V.; Hotchkiss, R.S. Interleukin 7 immunotherapy improves host immunity and survival in a two-hit model of Pseudomonas aeruginosa pneumonia. J. Leukoc. Biol. 2017, 101, 543–554.
- Francois, B.; Jeannet, R.; Daix, T.; Walton, A.H.; Shotwell, M.S.; Unsinger, J.; Monneret, G.; Rimmelé, T.; Blood, T.; Morre, M.; et al. Interleukin-7 restores lymphocytes in septic shock: The IRIS-7 randomized clinical trial. JCI Insight 2018, 3, e98960.
- Patera, A.C.; Drewry, A.M.; Chang, K.; Beiter, E.R.; Osborne, D.; Hotchkiss, R.S. Frontline Science: Defects in immune function in patients with sepsis are associated with PD-1 or PD-L1 expression and can be restored by antibodies targeting PD-1 or PD-L1. J. Leukoc. Biol. 2016, 100, 1239–1254.
- Zhang, Y.; Zhou, Y.; Lou, J.; Li, J.; Bo, L.; Zhu, K.; Wan, X.; Deng, X.; Cai, Z. PD-L1 blockade improves survival in experimental sepsis by inhibiting lymphocyte apoptosis and reversing monocyte dysfunction. Crit. Care 2010, 14, R220.
- Brahmamdam, P.; Inoue, S.; Unsinger, J.; Chang, K.C.; McDunn, J.; Hotchkiss, R.S. Delayed administration of anti-PD-1 antibody reverses immune dysfunction and improves survival during sepsis. J. Leukoc. Biol. 2010, 88, 233–240.
- Pierrakos, C.; Velissaris, D.; Bisdorff, M.; Marshall, J.C.; Vincent, J.-L. Biomarkers of sepsis: Time for a reappraisal. Crit. Care 2020, 24, 287.
- Wang, X.; Li, Z.-Y.; Zeng, L.; Zhang, A.-Q.; Pan, W.; Gu, W.; Jiang, J.-X. Neutrophil CD64 expression as a diagnostic marker for sepsis in adult patients: A meta-analysis. Crit. Care 2015, 19, 245.
- Bauer, P.R.; Kashyap, R.; League, S.C.; Park, J.G.; Block, D.R.; Baumann, N.A.; Algeciras-Schimnich, A.; Jenkins, S.M.; Smith, C.Y.; Gajic, O.; et al. Diagnostic accuracy and clinical relevance of an inflammatory biomarker panel for sepsis in adult critically ill patients. Diagn. Microbiol. Infect. Dis. 2016, 84, 175–180.
- Llewelyn, M.J.; Berger, M.; Gregory, M.; Ramaiah, R.; Taylor, A.L.; Curdt, I.; Lajaunias, F.; Graf, R.; Blincko, S.J.; Drage, S.; et al. Sepsis biomarkers in unselected patients on admission to intensive or high-dependency care. Crit. Care 2013, 17, R60.
- Monneret, G.; Debard, A.-L.; Venet, F.; Bohe, J.; Hequet, O.; Bienvenu, J.; Lepape, A. Marked elevation of human circulating CD4+CD25+ regulatory T cells in sepsis-induced immunoparalysis. Crit. Care Med. 2003, 31, 2068–2071.
- Matera, G.; Puccio, R.; Giancotti, A.; Quirino, A.; Pulicari, M.C.; Zicca, E.; Caroleo, S.; Renzulli, A.; Liberto, M.C.; Focà, A. Impact of interleukin-10, soluble CD25 and interferon-gamma on the prognosis and early diagnosis of bacteremic systemic inflammatory response syndrome: A prospective observational study. Crit. Care 2013, 17, R64.
- Doerflinger, M.; Reljic, B.; Menassa, J.; Nedeva, C.; Jose, I.; Faou, P.; Mackiewicz, L.; Mansell, A.; Pellegrini, M.; Hotchkiss, R.; et al. Circulating BiP/Grp78 is a novel prognostic marker for sepsis-mediated immune cell death. FEBS J. 2021, 288, 1809–1821.
- Polk, H.C., Jr.; George, C.D.; Wellhausen, S.R.; Cost, K.; Davidson, P.R.; Regan, M.P.; Borzotta, A.P. A systematic study of host defense processes in badly injured patients. Ann. Surg. 1986, 204, 282–299.
- Cheron, A.; Floccard, B.; Allaouchiche, B.; Guignant, C.; Poitevin, F.; Malcus, C.; Crozon, J.; Faure, A.; Guillaume, C.; Marcotte, G.; et al. Lack of recovery in monocyte human leukocyte antigen-DR expression is independently associated with the development of sepsis after major trauma. Crit. Care 2010, 14, R208.
- Li, J.; Tang, Z.; Xie, M.; Hang, C.; Yu, Y.; Li, C. Association between elevation of plasma biomarkers and monocyte dysfunction and their combination in predicting sepsis: An observational single-centre cohort study. Innate Immun. 2020, 26, 514–527.
- Monneret, G.; Venet, F. Monocyte HLA-DR in sepsis: Shall we stop following the flow? Crit. Care 2014, 18, 102.


