Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Julie Dardare | + 2341 word(s) | 2341 | 2021-08-04 03:10:24 | | | |
| 2 | Peter Tang | Meta information modification | 2341 | 2021-08-05 03:52:53 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Dardare, J. Epithelial to Mesenchymal Transition. Encyclopedia. Available online: https://encyclopedia.pub/entry/12783 (accessed on 10 August 2026).
Dardare J. Epithelial to Mesenchymal Transition. Encyclopedia. Available at: https://encyclopedia.pub/entry/12783. Accessed August 10, 2026.
Dardare, Julie. "Epithelial to Mesenchymal Transition" Encyclopedia, https://encyclopedia.pub/entry/12783 (accessed August 10, 2026).
Dardare, J. (2021, August 04). Epithelial to Mesenchymal Transition. In Encyclopedia. https://encyclopedia.pub/entry/12783
Dardare, Julie. "Epithelial to Mesenchymal Transition." Encyclopedia. Web. 04 August, 2021.
Copy Citation
Pancreatic ductal adenocarcinoma (PDAC) is one of the malignancies with the worst prognosis despite a decade of efforts. The effectiveness of PDAC therapies is challenged by the early and widespread metastasis. Epithelial to mesenchymal transition (EMT) is a major driver of cancer progression and metastasis.
PDAC
EMT
metastasis
biomarker
1. Introduction
Pancreatic cancer is the fourth-leading cause of death by cancer, and has the lowest 5-year relative survival rate (9%) as reported by The American Cancer Society reports in 2020 [1]. The incidence of pancreatic cancer continue to increase, and it is projected to become the second cause of cancer death before 2030 in Western countries [2]. The pancreatic ductal adenocarcinoma (PDAC) histological subtype represents almost 90% of pancreatic malignancies. Because of the lack of clinical symptoms in early stages and an high metastatic potential of PDAC cells, up to 80% patients are diagnosed at late stages [3]. The majority of the 15–20% patients eligible for a surgical resection finally relapse or develop a local or metastatic recurrence [3][4]. There is an urgent need to improve diagnosis with efficient prognostic biomarkers which would allow a better management of this disease.
Metastatic evolution remains a concern for the management of patients with PDAC. Epithelial to mesenchymal transition (EMT) is one of the key mechanisms that leads to tumor progression and development of metastasis [5][6]. EMT is a dynamic process in which epithelial cells loss their phenotype and acquire a mesenchymal phenotype. This transition is defined by loss of characteristic epithelial markers such as E-cadherin or cytokeratins and a gain of mesenchymal markers such as N-cadherin or vimentin [7]. These changes provide morphological modifications with remodeling of the cytoskeleton, disruption of cell adhesion capacity to other cells, and to the matrix, loss of cellular polarity. Taken together these events enhance invasiveness, migration and finally metastasis [5][6][8].
Two different EMT states have been described in PDAC in vivo models and define the cell dissemination type: partial EMT (pEMT) and complete EMT (cEMT) [9]. In pEMT, cells are stably or dynamically in an epithelial–mesenchymal intermediate state. Cells can express both epithelial and mesenchymal markers or they can loss epithelial features without a gain of mesenchymal features.
Furthermore, EMT has been identified as having a role at preneoplastic stage of PDAC in vivo, allowing cells to seeding to distant organs prior or in parallel to primary tumor formation [10]. Almost all patients with complete surgical resection and no metastasis finally die from disease within five years is consistent with this early spread model [3][11][12]
2. Epithelial to Mesenchymal Transition
EMT is a dynamic biological process in which epithelial cells evolves to a mesenchymal state. Epithelial cells normally interact with basement membrane through a basal-apical polarity, they are in close contact with each other through cell–cell junctions. During EMT, epithelial cells undergo changes in gene expression through multiple molecular processes leading to the repression of these epithelial characteristics and gain of mesenchymal features allowing cells migrative and invasive properties [13]. EMT is considered as a dynamic process because of its capacity to reverse the phenomenon through the mesenchymal–epithelial transition (MET), which is less understood. EMT plays crucial roles in the development and evolution of the disease: Three subtypes have been proposed: during implantation, embryogenesis, and organ development (type 1); during tissue regeneration and fibrosis (type 2); and associated with cancer progression and metastasis (type 3) [7].
During cancer progression, type 3 EMT allows carcinoma cells capacities to dissociate themselves from the primary tumor. Then, cells can disseminate through invasion, intravasation, survival in blood and lymphatic stream, extravasation, to finally develop a metastasis into secondary organs. Once at the metastatic site, cells that acquired mesenchymal-like phenotype through EMT seem to reverse the process (MET) to regain epithelial properties and to integrate into distant organs. This phenomenon can be illustrated by the fact that distant metastases are commonly composed of differentiated epithelial cells, however the implication of MET is still being debated [14].
Besides cellular migration, EMT plays a role in a myriad of processes implied in cancer pathways such as resistance to cell death, blocking senescence, enhancing survival, promoting genomic instability, metabolism modifications, drug resistance, and immune suppression [15]. Implication of EMT in cancer progression and metastasis appears to be different according to cancer type.
A new class of drugs termed “migrastatics” has been defined recently: These drugs interfere with all modes of cancer cell invasion and with their ability to metastasize [16]. Unlike conventional cancer drugs which target the proliferation of cancer cells, migrastatics focus on the inhibition of local invasion and metastasis. Recent work has developed a pipeline approach suitable for the development of migrastatics drugs in melanoma [17]. This theoretical approach might comprise EMT and might represent a promising new therapeutical strategy in cancer with high metastatic potential including PDAC.
2.1. EMT Signaling
Activation of EMT might be triggered by various signaling pathways depending on the tumor microenvironment. Indeed, tumor-associated stroma can increase the expression of EMT-transcription factors (EMT-TFs). Among signaling pathways are included transforming growth factor β (TGF-β), bone morphogenic protein (BMP), Notch, Wnt/β-catenin, sonic hedgehog, epidermal growth factor (EGF), fibroblast growth factor (FGF), and platelet-derived growth factor (PDGF) [18][19][20][21][22] (Figure 1). Activation of these EMT-inducing signaling pathways leads to the expression of transcription factors that governs EMT-associated genes. They simultaneously repressed the expression of epithelial genes, and on the other hand they induce genes associated with the mesenchymal phenotype. EMT-TFs include basic helix-loop-helix (bHLH) factors TWIST1 and TWIST2, the zinc finger E-box-binding homebox ZEB1 and ZEB2, and the zinc finger binding transcription factors SNAI1 and SNAI2.
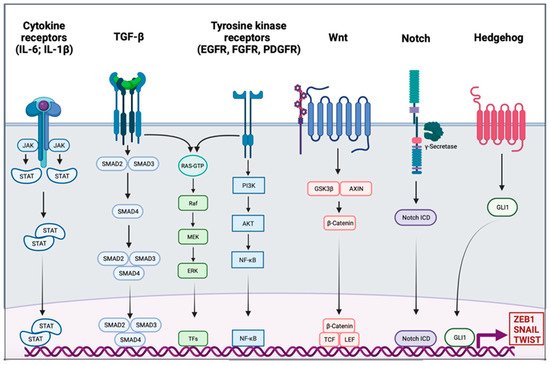
Figure 1. Signaling pathway involved in the epithelial to mesenchymal transition. Different signaling pathways can activate epithelial to mesenchymal transition (EMT) through the activation of EMT transcription factors ZEB1, SNAIL, and TWIST. Interleukin 6 (IL-6) and interleukin 1β (IL-1β) can bind cytokine receptors. Signaling is conducted through the activation of Janus kinase (JAK) and the recruitment of signal transducer and activator of transcription proteins (STATs); the dimer of STATs translocates into the nucleus to activate the transcription of genes. The transforming growth factor β (TGF-β) signal is conducted by SMADs protein into the nucleus, and the trimer activates the transcription. Tyrosine kinase receptors (RTK), such as epidermal growth factor receptor (EGFR), fibroblast growth factor receptor (FGFR), or platelet-derived growth factor receptor (PDGFR), induce PI3K, AKT, and nuclear factor-κB (NF-κB). The TGF-β pathway and RTK are also able to trigger the RAS-RAF-MEK-ERK signaling pathway. The WNT signaling results in the release of β-Catenin from the glycogen synthase kinase-3β (GSK3β)–axis inhibition protein (AXIN) complex. β-Catenin moves into the nucleus and binds to the transcription factors T cell factor (TCF) and the lymphoid enhancer-binding factor (LEF). Intracellular domain of the notch receptor (Notch ICD) is cleaved after the activation of the receptor, then it can translocate into the nucleus and act as a transcriptional co-activator. Hedgehog signaling induces EMT-associated gene expression through the activation of GLI1.
SNAIL is the first described transcriptional repressor of E-cadherin; it binds to the E box consensus sequence in the promoter of CDH1, encoding E-cadherin, and directly repress its transcription [23][24]. SNAIL also induces the downregulation of others epithelial molecules such as Claudins, Occludins, and Mucin-1. SNAIL also has the ability to directly induce mesenchymal genes such as Fibronectin and Matrix Metallopeptidase 9 (MMP9) [25]. ZEB1 and ZEB2 repress E-cadherin expression by directly binding to the E-Box element of CDH1 [26]. They also induce the expression of mesenchymal proteins such as N-cadherin and Vimentin [27][28]. Unlike the last two EMT-TFs, TWIST acts as an indirect repressor of E-cadherin partly due to its transcriptional activation of SNAI2 [29][30]. TWIST is also able to activate expression of mesenchymal genes such as N-cadherin and Vimentin [31]. The functional loss of E-cadherin is considered as a crucial step in EMT; however, many others epithelial proteins are also downregulated: Mucin-1, Occludins, Claudins, and Desmoplakin. On the other side, mesenchymal markers are gained, they include N-cadherin, Vimentin, Smooth Muscle Actin, Fibronectin, Matrix Metalloproteinases, and Vitronectin [32]. In addition to its role in transcriptional regulation, EMT might be orchestrated by other regulatory networks including regulation by microRNAs (miRs), differential splicing, translational and posttranslational control.
Molecular changes described above lead to cellular hallmarks of EMT including the loss of apical-basal polarity, disruption of cell-to-cell contacts (including adherent junctions, tight junctions, and desmosomes), cytoskeleton structure and ECM degradation by expressing matrix metalloproteinases. Consequently, cells ongoing EMT acquire a spindle-shape mesenchymal morphology which allows them motility and ability to degrade and invade their basal ECM [33].
3. EMT in PDAC
3.1. Activation of EMT in PDAC
In cancer cells, EMT can be activated through different stimuli. Marcucci et al. have identified five main classes of stimuli: mechanical stress, low pH and hypoxia, innate and adaptative immune responses, altered ECM and treatment with anti-tumor drugs [34]. PDAC is well known for its desmoplastic stroma, which is composed of a dense acellular extracellular matrix (ECM) infiltrated by heterogenous populations of immune, endothelial cells, and cancer associated fibroblasts (CAFs) [35]. A dense stroma can predispose the tumor microenvironment to limit delivery and diffusion of oxygen creating a hypoxic environment. The dense desmoplastic stroma composes a large part of the ECM and includes collagen, fibronectin, laminin, and hyaluronic acid. These ECM proteins are mainly produced by CAFs and in smaller amounts by cancer cells. They first have the ability to form a physical barrier, although they also can have signaling functions in EMT [36]. CAFs are able to produce various cytokines and chemokines such as TGF-β; interleukin 1 (IL-1); interleukin 6 (IL-6) and tumor necrosis factor α (TNFα), to the latter activating signaling pathways of EMT [36]. In PDAC cells, microenvironmental changes including hypoxia or TGF-β stimulation led to changes in EMT-markers within a decrease in E-cadherin and an increase in vimentin protein and mRNA levels [37] (Figure 2).
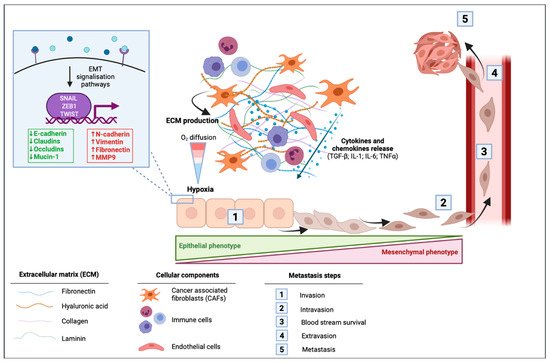
Figure 2. Impact of the tumor microenvironment of pancreatic ductal adenocarcinoma (PDAC) in the activation of epithelial to mesenchymal transition (EMT). Cancer-associated fibroblasts (CAFs) induce the production of extracellular matrix (ECM) that is composed of fibronectin, hyaluronic acid, collagen, and laminin. This dense desmoplastic stroma limits the diffusion of oxygen in the tumor and leads to hypoxia. CAFs release different cytokines and chemokines including the transforming growth factor β (TGF-β), interleukin 1 (IL-1), interleukin 6 (IL-6), and the tumor necrosis factor α (TNF-α). These extracellular mediators can activate signaling pathways leading to the activation of EMT in which cells switch from their epithelial phenotype to a mesenchymal phenotype with a spindle-shape morphology. EMT is then followed by intravasion of mesenchymal cells, blood stream survival, extravasion, and finally by the formation of metastasis.
Senescence and cancer are well known to be linked. In PDAC, senescence seems to occur in earliest stages and provides tumor suppressor effects. However, several evidences indicates that senescent cells in microenvironment can have a pro tumorigenic role in part with the senescence-associated secretory phenotype (SASP) [38]. Exposure to SASP can induce cell plasticity through the stimulation of cancer cell proliferation, motility and the generation of inflammatory environment [39]. Therefore, in PDAC microenvironment SASP could participates to enhance EMT.
EMT can also be favored by mutations, the major mutation found in PDAC in more than 90% of case, is the activation of the KRAS oncogene [40]. KRAS activation can modulate the tumor microenvironment and maintaining an active stroma through the production of IL-6 and sonic hedgehog, which play an important role in the EMT process [41]. Loss of SMAD4 is one of the fourth most common mutations found in PDAC, with an inactivation found in 60% of case, resulting in alterations in the TGF-β signaling pathway which is itself altered in 47% of PDAC cases [42]. Considering of the potential role of TGF-β in the induction of EMT, this alteration may have an impact during PDAC progression.
3.2. Role of EMT in PDAC Metastasis
EMT is traditionally considered as a binary phenomenon, allowing the transition from epithelial to mesenchymal phenotype, which is called “complete EMT” (cEMT). However, increased recent evidence suggests that cells undergoing EMT are heterogeneous and can express both epithelial and mesenchymal markers in a hybrid state called “partial EMT” (pEMT) [9][43][44]. Using a spontaneously metastatic genetically engineered mouse model (GEMMs) of PDAC, Aiello et al. found that EMT subtypes influence the mode of cell migration in metastasis. Tumor cells in cEMT subtype lacking E-cadherin protein preferentially disseminate as single cells. On the other hand, in the pEMT subtype, tumor cells expressing both epithelial and mesenchymal markers preferentially disseminate through collective migration [9]. EMT is a key step in metastasis which is a late stage of tumorigenesis. However, in pancreatic carcinomas EMT has been identified to occur at early stage. Rhim et al., assume that EMT occurs at preinvasive stages leading the seeding to distant organs to occur before and simultaneously to the primary tumor formation [10].
Despite clear evidences of EMT implication in tumor metastasis, the exact functions of EMT in cancer are still debated. Indeed, a study has challenged the role of EMT in metastasis, precisely on effects of EMT-TF SNAIL and TWIST in pancreatic cancer. Zheng et al. used the PDAC model KPC (Pdx1-cre; KRASG12D; p53R172H) in mouse in which they independently knockout TWIST or SNAIL. Despite inducing suppression of EMT, the loss of SNAIL or TWIST did not alter cancer progression or local invasion or metastasis. The authors therefore claim that EMT is dispensable for metastasis. In this study, EMT-TF knockout was also correlated with chemosensitivity to gemcitabine, which has been attributed to the increased expression of nucleotide transporters, authors conclude therefore EMT induces chemoresistance in pancreatic cancer [45]. Similar observations have been made in breast cancer for lung metastasis [46]. Krebs et al. used the same KPC mouse model with targeting another key EMT-TF ZEB1. In contrast to depletion in TWIST or SNAIL, ZEB1 knockout has impair multiple stages of tumorigenesis including precursor lesion formation, tumor grading, invasion, and metastasis that clearly demonstrate a key role of ZEB1 in the in vivo tumor progression of pancreatic cancer from premalignant lesion towards metastasis [47]. Previously another study using a short hairpin knockdown of ZEB1 in mice has also highlighted the importance of ZEB1 in tumor cells dissemination and in the tumor cells capacity to initiate tumor in pancreatic cancer [48]. Putting together these studies shows a trend to functional differences of EMT-TFs, although SNAIL and TWIST seem to be dispensable ZEB1 is on the contrary a key factor that appears to be not compensable by another EMT-TF.
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. A Cancer J. Clin. 2020, 70, 7–30.
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921.
- Ducreux, M.; Cuhna, A.S.; Caramella, C.; Hollebecque, A.; Burtin, P.; Goéré, D.; Seufferlein, T.; Haustermans, K.; Laethem, J.L.V.; Conroy, T.; et al. Cancer of the Pancreas: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up†. Ann. Oncol. 2015, 26, v56–v68.
- Lambert, A.; Schwarz, L.; Borbath, I.; Henry, A.; Van Laethem, J.-L.; Malka, D.; Ducreux, M.; Conroy, T. An Update on Treatment Options for Pancreatic Adenocarcinoma. Ther. Adv. Med. Oncol. 2019, 11, 1758835919875568.
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45.
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890.
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428.
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, Cell Plasticity and Metastasis. Cancer Metastasis Rev. 2016, 35, 645–654.
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695.
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell 2012, 148, 349–361.
- Lutfi, W.; Talamonti, M.S.; Kantor, O.; Wang, C.-H.; Liederbach, E.; Stocker, S.J.; Bentrem, D.J.; Roggin, K.K.; Winchester, D.J.; Marsh, R.; et al. Perioperative Chemotherapy Is Associated with a Survival Advantage in Early Stage Adenocarcinoma of the Pancreatic Head. Surgery 2016, 160, 714–724.
- Strobel, O.; Neoptolemos, J.; Jäger, D.; Büchler, M.W. Optimizing the Outcomes of Pancreatic Cancer Surgery. Nat. Rev. Clin. Oncol. 2019, 16, 11–26.
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and Definitions for Research on Epithelial-Mesenchymal Transition. Nat. Rev. Mol. Cell Biol 2020, 21, 341–352.
- Polyak, K.; Weinberg, R.A. Transitions between Epithelial and Mesenchymal States: Acquisition of Malignant and Stem Cell Traits. Nat. Rev. Cancer 2009, 9, 265–273.
- Bhatia, S.; Wang, P.; Toh, A.; Thompson, E.W. New Insights Into the Role of Phenotypic Plasticity and EMT in Driving Cancer Progression. Front. Mol. Biosci 2020, 7, 71.
- Gandalovičová, A.; Rosel, D.; Fernandes, M.; Veselý, P.; Heneberg, P.; Čermák, V.; Petruželka, L.; Kumar, S.; Sanz-Moreno, V.; Brábek, J. Migrastatics—Anti-Metastatic and Anti-Invasion Drugs: Promises and Challenges. Trends in Cancer 2017, 3, 391–406.
- Maiques, O.; Fanshawe, B.; Crosas-Molist, E.; Rodriguez-Hernandez, I.; Volpe, A.; Cantelli, G.; Boehme, L.; Orgaz, J.L.; Mardakheh, F.K.; Sanz-Moreno, V.; et al. A Preclinical Pipeline to Evaluate Migrastatics as Therapeutic Agents in Metastatic Melanoma. Br. J. Cancer 2021.
- Xu, J.; Lamouille, S.; Derynck, R. TGF-Beta-Induced Epithelial to Mesenchymal Transition. Cell Res. 2009, 19, 156–172.
- McCormack, N.; O’Dea, S. Regulation of Epithelial to Mesenchymal Transition by Bone Morphogenetic Proteins. Cell Signal. 2013, 25, 2856–2862.
- Espinoza, I.; Miele, L. Deadly Crosstalk: Notch Signaling at the Intersection of EMT and Cancer Stem Cells. Cancer Lett. 2013, 341, 41–45.
- Taipale, J.; Beachy, P.A. The Hedgehog and Wnt Signalling Pathways in Cancer. Nature 2001, 411, 349–354.
- Gonzalez, D.M.; Medici, D. Signaling Mechanisms of the Epithelial-Mesenchymal Transition. Sci. Signal. 2014, 7, re8.
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; García De Herreros, A. The Transcription Factor Snail Is a Repressor of E-Cadherin Gene Expression in Epithelial Tumour Cells. Nat. Cell Biol. 2000, 2, 84–89.
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The Transcription Factor Snail Controls Epithelial-Mesenchymal Transitions by Repressing E-Cadherin Expression. Nat. Cell Biol. 2000, 2, 76–83.
- Wu, Y.; Zhou, B.P. Snail: More than EMT. Cell Adh. Migr. 2010, 4, 199–203.
- Eger, A.; Aigner, K.; Sonderegger, S.; Dampier, B.; Oehler, S.; Schreiber, M.; Berx, G.; Cano, A.; Beug, H.; Foisner, R. DeltaEF1 Is a Transcriptional Repressor of E-Cadherin and Regulates Epithelial Plasticity in Breast Cancer Cells. Oncogene 2005, 24, 2375–2385.
- Vandewalle, C.; Comijn, J.; De Craene, B.; Vermassen, P.; Bruyneel, E.; Andersen, H.; Tulchinsky, E.; Van Roy, F.; Berx, G. SIP1/ZEB2 Induces EMT by Repressing Genes of Different Epithelial Cell–Cell Junctions. Nucleic Acids Res. 2005, 33, 6566–6578.
- Bindels, S.; Mestdagt, M.; Vandewalle, C.; Jacobs, N.; Volders, L.; Noël, A.; van Roy, F.; Berx, G.; Foidart, J.-M.; Gilles, C. Regulation of Vimentin by SIP1 in Human Epithelial Breast Tumor Cells. Oncogene 2006, 25, 4975–4985.
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a Master Regulator of Morphogenesis, Plays an Essential Role in Tumor Metastasis. Cell 2004, 117, 927–939.
- Casas, E.; Kim, J.; Bendesky, A.; Ohno-Machado, L.; Wolfe, C.J.; Yang, J. Snail2 Is an Essential Mediator of Twist1-Induced Epithelial Mesenchymal Transition and Metastasis. Cancer Res. 2011, 71, 245–254.
- Lamouille, S.; Xu, J.; Derynck, R. Molecular Mechanisms of Epithelial–Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196.
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of Epithelial-Mesenchymal Transition through Epigenetic and Post-Translational Modifications. Mol. Cancer 2016, 15, 18.
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374.
- Marcucci, F.; Stassi, G.; De Maria, R. Epithelial-Mesenchymal Transition: A New Target in Anticancer Drug Discovery. Nat. Rev. Drug Discov. 2016, 15, 311–325.
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The Pancreas Cancer Microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276.
- Bulle, A.; Lim, K.-H. Beyond Just a Tight Fortress: Contribution of Stroma to Epithelial-Mesenchymal Transition in Pancreatic Cancer. Signal. Transduct. Target. Ther. 2020, 5, 249.
- Wang, W.; Dong, L.; Zhao, B.; Lu, J.; Zhao, Y. E-cadherin Is Downregulated by Microenvironmental Changes in Pancreatic Cancer and Induces EMT. Oncol. Rep. 2018, 40, 1641–1649.
- Porciuncula, A.; Hajdu, C.; David, G. The Dual Role of Senescence in Pancreatic Ductal Adenocarcinoma. Adv. Cancer Res. 2016, 131, 1–20.
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The Senescence-Associated Secretory Phenotype Induces Cellular Plasticity and Tissue Regeneration. Genes Dev. 2017, 31, 172–183.
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.
- di Magliano, M.P.; Logsdon, C.D. Roles for KRAS in Pancreatic Tumor Development and Progression. Gastroenterology 2013, 144, 1220–1229.
- Dardare, J.; Witz, A.; Merlin, J.-L.; Gilson, P.; Harlé, A. SMAD4 and the TGFβ Pathway in Patients with Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2020, 21.
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226.
- Grigore, A.D.; Jolly, M.K.; Jia, D.; Farach-Carson, M.C.; Levine, H. Tumor Budding: The Name Is EMT. Partial EMT. J. Clin. Med. 2016, 5, 51.
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-Mesenchymal Transition Is Dispensable for Metastasis but Induces Chemoresistance in Pancreatic Cancer. Nature 2015, 527, 525–530.
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. EMT Is Not Required for Lung Metastasis but Contributes to Chemoresistance. Nature 2015, 527, 472–476.
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-Activator Zeb1 Is a Key Factor for Cell Plasticity and Promotes Metastasis in Pancreatic Cancer. Nat. Cell Biol. 2017, 19, 518–529.
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-Activator ZEB1 Promotes Tumorigenicity by Repressing Stemness-Inhibiting MicroRNAs. Nat. Cell Biol. 2009, 11, 1487–1495.
More
Information
Subjects:
Pharmacology & Pharmacy
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
2 times
(View History)
Update Date:
05 Aug 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No