Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mario García-Domínguez | + 6839 word(s) | 6839 | 2021-08-03 07:42:47 | | | |
| 2 | Camila Xu | -2 word(s) | 6837 | 2021-08-05 04:26:00 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
García-Domínguez, M. BET Family Proteins. Encyclopedia. Available online: https://encyclopedia.pub/entry/12771 (accessed on 10 July 2026).
García-Domínguez M. BET Family Proteins. Encyclopedia. Available at: https://encyclopedia.pub/entry/12771. Accessed July 10, 2026.
García-Domínguez, Mario. "BET Family Proteins" Encyclopedia, https://encyclopedia.pub/entry/12771 (accessed July 10, 2026).
García-Domínguez, M. (2021, August 04). BET Family Proteins. In Encyclopedia. https://encyclopedia.pub/entry/12771
García-Domínguez, Mario. "BET Family Proteins." Encyclopedia. Web. 04 August, 2021.
Copy Citation
The BET family of proteins consists of a series of proteins with two N-terminal tandem bromodomains and an exclusive extra terminal domain (ET) that play an important role in gene transcription through epigenetic regulation, with a prominent impact in the control of cell growth and differentiation.
SARS-CoV-2
COVID-19
BET
BET inhibitors
BRD4
BRD2
virus
immunity
inflammation
1. BET Proteins
The BET family of proteins consists of a series of proteins that play an important role in gene transcription through epigenetic regulation, with a prominent impact in the control of cell growth and differentiation [1][2][3][4]. In mammals, the BET family is composed of four members: BRD2, BRD3, BRD4 and BRDT (Figure 1). While BRD2, BRD3 and BRD4 expression occurs ubiquitously, BRDT expression is restricted to the male germline [5]. Recently, BRD2 and BRD4 were reported to interact with the envelope (E) protein of SARS-CoV-2 [6], which makes of these BET members potential targets for host-directed therapy strategies. Moreover, several groups have demonstrated that angiotensin-converting enzyme 2 (ACE2), the main SARS-CoV-2 receptor for host cell entry, is under BET protein transcriptional regulatio [7][8][9][10].
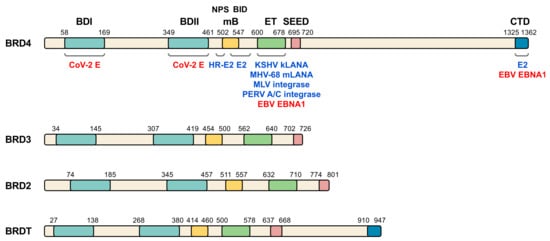
Figure 1. BET family of proteins and viral interacting proteins. The four members of human BET family are represented and amino acid position of the main relevant domains is indicated (BDI, bromodomain I; BDII, bromodomain II; mB, motif B; ET, extra terminal domain; SEED, SEED domain; CTD, C-terminal domain; NPS, N-terminal phosphorylation sites region; BID, basic residue-enriched interaction domain). Different virus proteins demonstrated to directly interact with BET domains (blue) or postulated to interact (red) are shown on BRD4, although some of the indicated proteins also interact with other BET members. CoV-2 E, SARS-CoV-2 E protein. See Table 1 and text for details and references.
Table 1. BET interactions with viruses.
| Virus | Viral Protein/Genome | BET | BET Domain | Functions | Refs. |
|---|---|---|---|---|---|
| Papillomavirus HR-HPV |
E2 HR-E2 |
BRD4 | CTD, BID CTD, BID, NPS |
E2 stability, E2-mediated transcription, E2 tethering to mitotic chromatin | [11][12][13] |
| KSHV | kLANA | BRD2, BRD3, BRD4 |
ET | kLANA tethering to chromatin and TSSs | [14][15][16][17][18] |
| MHV-68 | mLANA | BRD2, BRD3, BRD4 |
ET | mLANA tethering to chromatin and TSSs | [18][19] |
| EBV | EBNA1 OriLyt ** |
BRD2, BRD3, BRD4 |
(ET, CTD) * (BDs) |
EBNA1-mediated transcription Late gene expression |
[20][21] |
| MLV | integrase | BRD2, BRD3, BRD4 |
ET | Integration into TSSs and CpG islands | [22][23][24][25] |
| PERV A/C | integrase | BRD2, BRD3, BRD4 |
ET | Integration cofactor | [26] |
| HIV | Tat *** LTR |
BRD4 | CTD BDs |
Competence for P-TEFb HIV transcription and latency |
[27][28][29][30] |
| HTLV-1 | Tax | BRD4 | CTD | Competence for P-TEFb | [31] |
| HCMV | Promoters | BRD4 | CTD | Competence for P-TEFb for transcription | [32] |
| SARS-CoV-2 | E | BRD2, BRD4 |
(BDs) | (Transcription) | [6] |
* brackets indicate putative interaction domains or function. ** italic indicates virus genome regions. *** underlining indicates non-direct interaction but competition for P-TEFb.
A salient feature of this family of proteins is the presence of two tandem N-terminal bromodomains able to bind acetyl groups. Interaction with histones through acetyl-group recognition on lysine (K) residues constitutes the main mechanistic aspect of BET action. The tight relation of BET proteins with cell cycle progression explains why they are linked to many cancer types. This implication was initially illustrated in the context of fusions of BET members with the NUclear protein of the Testis (NUT), which gives rise to NUT midline carcinoma [2], but misregulation of BET expression is in the basis of many other types of cancer (reviewed in [33]). Thus, the development of drugs able to displace BET proteins from the chromatin as a therapeutic approach to fight cancer has been a highly active research objective in the last decade (reviewed in [34]). Besides, BET inhibition has also proven to be of interest for treating metabolic, cardiovascular, neurological and autoimmune diseases (reviewed in [35][36][37]). The inhibitory strategy has been long based on synthetic drugs mimicking the acetyl-lysine group, thus able to compete with chromatin for BET binding and to displace these from target sites. This approach successfully alleviates a number of cancer types in mouse and cellular models [38][39][40][41][42]. However, success in clinical trials with humans is limited, probably due to the toxicity of the high doses required for effective outcomes [34][43]. Of note, thrombocytopenia is among the most common and severe adverse events associated with BET inhibition [44]. Besides, despite the high selectivity of BET inhibitors for BET bromodomains, 44 additional human proteins have bromodomains [45], making it difficult to completely discount off-target effects.
Though BET inhibitors have limited success in the treatment of some cancers, recent reports have shown BET inhibition as a promising strategy for treating SARS-CoV-2 infection [7][8][9][10]. Thus, BET inhibition anticipates as a solid host-directed therapy against COVID-19. Strikingly, BET proteins are tightly linked to infection by other viruses. BET relation with viruses occurs at two levels. On the one hand, BET proteins are targets for several virus proteins, which may result in an impact on cell transcription [46]. On the other hand, BET proteins are involved in activating transcriptional programs related to immunity and the inflammatory response associated with infection [47][48].
1.1. BET Structure
BET family of proteins is characterized by a common domain structure (Figure 1), presenting from N- to C-terminus: the two bromodomains (BDI and BDII), a motif B, a well conserved extra-terminal (ET) domain and a less conserved domain called SEED due to the presence of serine and glutamic and aspartic acid residues [49]. Bromodomains comprise a conserved sequence of approximately 110 amino acids [50][51] that can bind to acetylated lysine residues in histones and other proteins, like the GATA-1 transcription factor [52][53][54]. These domains, through their interaction with nucleosomes in chromatin, are involved in epigenetic regulation of gene transcription [55]. Of note, BET bromodomains have been suggested to be the target of SARS-CoV-2 E protein [6]. Major histone recognition by BETs relies on acetylation of K5 and K12 on histone H4 [4][56][57][58][59][60][61][62][63]. Interestingly, SARS-CoV-2 non-structural protein (nsp) 5 interacts with histone deacetylase 2 (HDAC2) [6], leading to speculation about interference of SARS-CoV-2 with histone acetylation. The motif B presents a coiled coil structure that gives rise to an amphipathic helix and is an essential domain for homo- and heterodimerization of BET members, thus stabilizing its binding to the chromatin and facilitating its association to chromosomes during mitosis [64]. This region also contributes to partner recruitment [65][66]. The ET domain consists of a region of approximately 80 amino acids and is involved in the interaction with specific effector proteins [67][68]. Of note, the ET domain is the major target of most viral proteins interacting with members of the BET family [46] (Figure 1). For its part, both BRD4 and BRDT have a C-terminal domain (CTD) at their carboxyl extreme whose main function is to interact with the positive transcription elongation factor b (P-TEFb) [55][27][69]. Importantly, virus infection frequently associates with altered host transcription. In this context, the analysis of transcriptional profiles in cellular and animal models of SARS-CoV-2 infection and in patient samples, have permitted to identify unique transcriptional signature in response to the virus [70].
1.2. General BET Functions
A variety of reports link BET proteins with cell cycle progression. BRD2 interacts with and activates E2F, a transcription factor involved in the synthesis of proteins required for the G1 to S transition during the cell cycle [71][72], proving to be involved in the control of Ccna2 (Cyclin A2) and Ccnd1 (Cyclin D1) genes [59][73]. Initial reports on BRD4 suggested its participation in the G2 to M transition [74], but more recent reports have solidly established that it is required for the M to G1 transition [75][76]. On its hand, BRDT is required for expression of Ccna1 (Cyclin A1) during spermatogenesis [77][78]. Importantly, SARS-CoV-2 nsp1, which is involved in inhibition of host protein expression, has been shown to induce cell cycle arrest in G0/G1 phase [79].
BRD4 was shown to be associated with chromosomes in interphase but also to remain bound to chromosomes during mitosis, when most nuclear regulatory factors are released into the cytoplasm [56][74]. Attachment to mitotic chromosomes has also been reported for BRD2 [64]. These observations, together with BET requirement for cell cycle progression, have led to consider BET proteins as true epigenetic factors, marking key chromatin positions from one generation to the next for timely activation of relevant cell cycle genes [80].
Several lines of evidence illustrate the role of BET proteins in organizing and maintaining the chromatin structure. BRDT has been shown to be a chromatin organizer in male germinal cells [3] and it was early observed that ectopic expression in somatic cells leads to dramatic reorganization of the chromatin [81]. In the same line, interfering with BRD4 leads to chromatin decondensation and fragmentation [82]. On its part BRD2 has been shown to cooperate with CTCF to enforce transcriptional and architectural boundaries at chromatin [83]. BET proteins act as histone chaperones of the acetylated nucleosomes they recognize, allowing the passage of RNA polymerase II to elongate nascent transcripts [59]. As mentioned, the CTD in BRD4 and BRDT is crucial for interaction with P-TEFb [55][27][69][84]. Interestingly, several viral proteins have been shown to compete with BRD4 for P-TEFb (see Section 3). BRD4 releases P-TEFb from inhibition by HEXIM1 [84]. Active P-TEFb phosphorylates Ser2 of the RNA Polymerase II C-terminal motif promoting RNA polymerase transcription elongation [85][86][87][88]. Recently, it has been described as a type of regulatory elements, the “super-enhancers” (SEs), which consist in large clusters of enhancers arranged in a cell type-specific manner [89][90]. They have emerged as key disease drivers when dysregulated, especially in the oncogenic transformation [91]. These elements represent a small fraction of the total enhancers in a cell, but they recruit a large proportion of regulatory proteins, strikingly BRD4, for the control of specific genes involved in the maintenance of cell identity. Thus, altered regulation of SEs may lead to changes in cell identity and cellular transformation. Indeed, cancer cells become highly dependent on these regulatory elements, so their specific targeting appears as a promising avenue for therapeutic intervention [92].
BET proteins are essential for development. Knock out mice for Brd4 and Brd2 die at early (postimplantation) and later (E11.5–E13.5) embryonic stages, respectively [93][94][95][96]. Heterozygous mice for these mutations also present defects, especially reduced cell growth. Particular association of BET proteins with development of the nervous system has been indicated. On the one hand, a BRD2 deficit is associated with defects in the developing neural tube, where the gene is highly expressed [93][95], and with a decrease in the number of GABAergic neurons [97]. On the other hand, BRD4 regulates the transcription of genes involved in synapses, enhancing learning and memory processes in mice [98]. On its hand, BRDT is well documented to play a prominent role in spermatogenesis [3][78].
2. BET Relation with Viruses
BET protein involvement in viral processes is not restricted to SARS-CoV-2. Abundant literature illustrates how BET proteins are strongly associated with infection by different types of viruses [46][48]. While some viral proteins directly interact with members of the BET family, others interfere with functional BET partners. The papillomaviruses E2 protein, the latency-associated nuclear antigen (LANA) of some herpesvirus, the integrase of some gamma-retroviruses and the E protein of SARS-CoV-2 are examples of viral proteins physically interacting with BET proteins [6][46], while human immunodeficiency virus type 1 (HIV-1) Tat or human T-lymphotropic virus type 1 (HTLV-1) Tax proteins can compete with BETs for PTEF-b [46]. Here, we summarize BET relation with different viruses, including SARS-CoV-2 (Figure 1 and Table 1), which outlines different strategies in viral infection leading to diverse scenarios for therapeutic BET inhibition.
The papillomaviruses E2 protein is essential for transcriptional activation and repression [99][100], for viral DNA replication in cooperation with the E1 protein [99] and for tethering of the viral genome to host mitotic chromosomes [101]. It was observed in bovine papillomavirus that BRD4 colocalizes with E2 in the mitotic chromatin and that E2 interact with the CTD of BRD4 [11]. BRD4 binding to E2 prevents E2 degradation, modulates E2-mediated transcription and tethers E2 to mitotic chromatin [12], despite some examples of E2 proteins associating with mitotic chromosomes in a BRD4 independent manner [102]. However, what has been undoubtedly shown is that BRD4 plays a dual role in regulating the transcriptional function of E2 proteins in papillomaviruses. Meanwhile different studies using diverse approaches show that BRD4 is essential for the activation of E2 transcriptional function [102][103][104][105][106], others show that BRD4 confers the ability to silence E2-mediated transcription [107][108]. Similarly, there are studies that attribute an essential role to BRD4 in viral genome replication [109], while others indicate that its role is not essential [110]. The most widely accepted current model contemplates that after infection of the cell by papillomavirus, BRD4 tethers the viral genome to active cellular chromatin to allow viral transcription. After binding to chromosomes, it recruits E1 and E2. As the genome replicates and the foci enlarge, BRD4 appears not to be required for continued replication of the genome (reviewed in [111]). More recent studies have shown that the interaction between the E2 protein and BRD4, besides occurring at the level of the CTD, also occurs at a basic residue-enriched interaction domain (BID). Moreover, it has been indicated that high-risk (HR) human papillomaviruses (HPV), associated with cervical cancer, but not low-risk (LR) HPV, associated with benign lesions of the genital tract [112], additionally interact with an N-terminal phosphorylation sites region (NPS) of BRD4 in a phosphorylation-dependent manner [13].
Kaposi’s sarcoma associated herpesvirus (KSHV), which causes Kaposi’s sarcoma, primary effusion lymphoma and some forms of Castleman’s disease, murine gamma-herpesvirus 68 (MHV-68) and the gamma-herpesvirus Epstein Barr virus (EBV), are also known to interact with BET proteins [19][20][113][114][115]. The LANA of the KSHV (kLANA) was the first viral protein discovered to interact with a member of the BET family protein [14]. kLANA plays important roles in replication [116][117][118][119][120], tethering of viral genome to cellular chromosomes [121][122][123][124][125][126][127] and regulation of viral and cellular gene transcription [116][117][128][129][130][131][132][133]. Its homologue in MHV-68, mLANA, is expressed in latency and during lytic replication and its function is essential in the establishment and maintenance of latency [134][135][136][137][138][139] and the EBV homologue, the Epstein-Barr virus nuclear antigen 1 (EBNA1), is involved in the regulation of viral transcription, replication and persistence [140]. Several studies show that kLANA and mLANA interact with a region containing the ET domain of BRD2, BRD3 and BRD4 [14][15][16][17][19], and a recent study has described that the preferential localization of kLANA and mLANA at transcription start sites (TSSs) [141][142][143][144] is due in part to BET proteins [18]. In this work, authors have shown that treatment with the BET inhibitor I-BET151 displaces kLANA protein, as well as BRD2 and BRD4, from viral and host TSSs. Mutations in mLANA preventing BRD2 and BRD4 binding [145] also have similar consequences [18]. In turn, EBNA1 has been described to interact with BRD4 and this interaction appears to play a critical role in EBNA1-mediated transcriptional activation, as it has been shown that BRD4 silencing leads to a decrease in its transcriptional activity [20]. In addition, it has been reported that BET inhibition on the one hand prevents expression of the viral immediate-early protein BZLF1, but on the other hand it also prevents viral late gene expression, as BET members localize and act on lytic origins of replication [21]. A different relationship with BET proteins has been attributed to other herpesvirus, where no physical interaction occurs. This is the case for human cytomegalovirus (HCMV), being BET proteins pivotally connected to the regulation of cytomegalovirus latency and reactivation. During latency, P-TEFb remains sequestered due to its interaction with BRD4, which prevents transcription of viral genes. Treatment with BET inhibitors allows the release of P-TEFb and thus the transcription of HCMV lytic genes, thanks to its recruitment to the promoters of the super elongation complex. But all this occurs without inducing viral DNA replication and complete reactivation [32].
Regarding retroviruses, the murine leukemia virus (MLV) integrase is also known to interact with the ET domain of BRD2, BRD3 and BRD4 [22][23][24][25]. Similarly, the porcine endogenous retrovirus A/C (PERV A/C) integrase interacts with the ET domain of BET proteins. This integrase protein interaction appears to be gamma-retrovirus specific, as BRD2 does not interact with either Rous associated virus type 1 (RAV-1, alpha-retrovirus) or HIV-1 (lentivirus) integrases [26]. In the case of the HIV, BRD4 plays an important role in transcriptional regulation [27][28][29][146]. On the one hand, the Tat protein of HIV is essential for transcriptional elongation from the long terminal repeat (LTR) promoter and several studies have shown that BRD4 inhibits the transcriptional elongation by competing with Tat for cellular P-TEFb [27][28][29]. Therefore, inhibition of BRD4 expression would lead to increased Tat-mediated HIV gene transcription. But on the other hand, BRD4, through its bromodomains can also be recruited to the HIV LTR by interacting with acetylated histones H3 and H4. The effects on HIV transcription and latency establishment are different depending on which of the histones it interacts with [30]. Treatment with the BET inhibitors JQ1, apabetalone, PFI-1 or UMB-136, has proven to reactivate latent HIV, helping to eradicate the virus [28][29][147][148]. However, treatment with the small molecule ZL0580, has shown suppression of HIV induction by establishing a more repressive chromatin structure at the HIV LTR, as well as by inhibiting Tat-mediated transcription transactivation and elongation [149]. For its part, the retrovirus HTLV-1 encodes the Tax protein which plays an important role in viral replication, transformation and transcriptional activation [150][151][152][153][154][155][156]. Similar to HIV Tat, Tax protein competes with BRD4 for binding to P-TEFb, which is essential for LTR promoter transactivation by Tax. Therefore, HTLV-1 Tax protein, like HIV Tat protein, appears to mimic the function of BRD4 and competes with it for binding to P-TEFb [31]. Interestingly, inhibition of BRD4 with JQ1 results in impaired proliferation of Tax-positive HTLV-1-infected cells, and then, in reduced Tax-mediated cell transformation and tumorigenesis [157], suggesting that BET inhibitors could also be used as anti-cancer therapy in tumors caused by viral infections.
BRD4 inhibition leads to viral arrest in infection by different types of viruses. In cells infected with pseudorabies virus (PRV), herpes simplex virus type 1 (HSV1), ectromelia virus (ECTV), vesicular stomatitis virus (VSV), porcine reproductive and respiratory syndrome virus (PRRSV), Newcastle disease virus (NDV) and influenza virus (H1N1), inhibition of BRD4 with different drugs (JQ1, OTX-015 and I-BET151) has shown antiviral activity. In a number of cases no changes in viral transcription was observed upon BRD4 inhibition, but an attenuation of viral attachment [158].
It has also been shown that respiratory syncytial virus (RSV) infection affects the BRD4 interactome, increasing the interaction with transcription factors involved in the innate immune response and cellular stress, and that this recruitment of multiple transcription factors occurs in a manner dependent on acetyl-lysine recognition [159].
As mentioned, it was recently reported the interaction of the SARS-CoV-2 E protein with BRD2 and BRD4 [6]. In this work, it is indicated that the histone H2A N-terminal tail shares similarity with a region of about 15 amino acids of E protein. The indicated histone region contains the target K residues for acetylation recognized by BET members. This observation has led to suggest the involvement of the BET bromodomains in the interaction with E protein, and to speculate about E protein ability to disrupt BET interaction with chromatin, which may affect host transcription in benefit of the virus [6]. However, the main target domains for interaction with viral proteins on BET members are the ET and CTD domains. Thus, interaction of E protein with bromodomains is unexpected and needs confirmation.
3. BET Proteins and SARS-CoV-2 Infection
Viruses need to use the machinery of the cells they infect to replicate. As explained, first evidence of the SARS-CoV-2-BET relation resulted from a proteomic study by Gordon et al. revealing the interaction of the SARS-CoV-2 E protein with BRD2 and BRD4 [6]. As BET proteins are general transcriptional co-regulators, Gordon et al. suggested that BET interaction with E protein may cause gene expression changes in the host cell that could be beneficial to the virus cycle. Besides being involved in cell cycle progression, BET proteins are also relevant for regulation of immunity and inflammation. Indeed, one decade ago, the pioneering BET drug I-BET was shown to displace BET proteins from regulatory elements on key inflammatory genes, displaying then anti-inflammatory properties [160]. Recent works have revealed that the beneficial use of BET inhibitors against SARS-CoV-2 is beyond the BET-E protein interaction.
Detection of viral antigens leads to antigen presentation to natural killer cells and CD8+ T-cells, which activate the immune response by production of proinflammatory cytokines and chemokines [161]. Normally, viruses are first faced by the innate immune response, but efficient counteraction may also require mobilization of the adaptive immune response (antigen targeting by immune cells). SARS-CoV-2 infection is linked to both responses [162]. However, BETs, and especially BRD4, are mainly involved in the innate immune response (reviewed in [48]). Production of proinflammatory cytokines is highly dependent on the Nuclear Factor kappa-light-chain-enhancer of activated B cells (NF-ĸB) signaling pathway [163]. Cytokines are essential for the immune response, but their aberrant dysregulation leads to hyperinflammation and may cause severe damage to tissues resulting in organ failure and death. This uncontrolled systemic inflammatory response is known as cytokine storm (CS) [164] and may account for up to 5% of COVID-19 patients [165]. Indeed, over-stimulation of the immune response can be more damaging than the virus infection itself. ACE2 is involved in cleaving angiotensin II into anti-inflammatory angiotensins 1–7. Thus, it has been suggested that ACE2 blocking by SARS-CoV-2 binding should result in accumulation of pro-inflammatory angiotensin II, contributing to the exacerbated immune response [166]. However, uncontrolled inflammation might also rely on pre-inflammatory states of certain organs and/or tissues.
3.1. SARS-CoV-2
Coronaviruses belong to the subfamily Orthocoronavirinae of the family Coronaviridae in the order Nidovirales. They are highly diverse, enveloped, positive-sense and single-stranded RNA viruses [167]. According to their genome structure and phylogenetic relationships, there are four genera of coronaviruses: alpha-, beta-, gamma- and delta-coronaviruses. While gamma- and delta-coronaviruses infect birds and some mammals, alpha- and beta-coronaviruses are responsible for the infection of various types of mammals [168], causing respiratory disorders in humans and gastroenteritis in other animals [169][170]. SARS-CoV-2 belongs to the beta-coronaviruses and is the seventh member of the coronavirus family to cause infections in humans [171]. Among coronaviruses infecting humans we find several common cold viruses like HCoV-OC43, HCoV-HKU1 and HCoV-229E [172], but coronaviruses with a high pathogenic capacity in humans have emerged in the last two decades, including SARS-CoV in 2002 and 2003 which caused 8000 confirmed cases with a 10% fatality rate, and MERS-CoV in 2012 with 2500 cases and a death rate of 36% [173]. Analysis of SARS-CoV-2 genome revealed 79.5% genomic identity with SARS-CoV [170].
The SARS-CoV-2 genome of approximately 30 kb encodes 14 open reading frames (ORFs) (Figure 2A). The ORF1ab is located in the 5′ region and encodes the overlapping polyproteins 1ab and 1a, which undergo auto-proteolysis to give rise to 16 non-structural proteins (nsps), mostly contributing to the formation of the replicase/transcriptase complex (RTC). At the 3′ end up to 13 ORFs are present, which include four structural proteins (Figure 2B): Spike (S), Envelope (E), Membrane (M) and Nucleocapsid (N) and nine putative accessory factors [174][175].
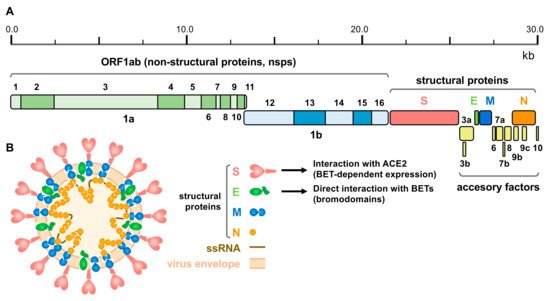
Figure 2. SARS-CoV-2 genomic structure and proteins. (A) Schematic representation of the SARS-CoV-2 genome, indicating the different derived proteins. ORF 1ab give rise to two polyproteins for non-structural proteins: pp1a and pp1ab, this last through a programmed ribosomal frameshift. Structural proteins comprise: Spike (S), Envelope (E), Membrane (M) and Nucleocapsid (N) proteins. (B) Schematic representation of the viral particle showing the viral envelope, the single strand RNA (ssRNA) genome and structural proteins (proteins are not represented to scale). Interaction of S protein with ACE2 (under the control of BET proteins) and direct interaction of E protein with BETs (putatively involving the bromodomains) is also indicated.
The N protein binds to the viral genome and is involved in RNA replication, virion formation and immune evasion, and also interacts with the M protein [176]. The M protein promotes viral particle assembly and budding through interaction with the N protein and accessory proteins 3a and 7a [161][177]. The most relevant structural proteins for BET inhibition are E and S. E protein has been demonstrated to interact with BRD2 and BRD4 [6]. It is the smallest structural protein and facilitates the production, maturation and release of virions [178]. S protein is a transmembrane protein that facilitates the binding of the viral envelope to the host receptor. The main receptor of SARS-CoV-2 in host cells is ACE2 [170][179], which is expressed in the colon, gallbladder, heart, kidney, epididymis, breast, ovary, lung, prostate, esophagus, tongue, liver, pancreas, and cerebellum [180]. Similar to other coronaviruses, SARS-CoV-2 needs to proteolyze S protein to activate the endocytic pathway. Host proteases, including transmembrane protease serine 2 (TMPRSS2), cathepsin L and furine have been shown to be involved in the process [179][181][182]. TMPRSS2 is highly expressed in certain tissues and co-expressed with ACE2 in bronchial branches, lungs and nasal epithelial cells, which explains part of the tissue tropism of SARS-CoV-2 [183][184]. Notably, expression of ACE2, but also of TMPRSS2, is under the control of BET proteins, what has been exploited to fight SARS-CoV-2 infection [7][8][9][10] (see below).
Of the 4 structural proteins, SARS-CoV-2 shares more than 90% amino acid identity with SARS-CoV, except for the S protein, which presents greater divergence [170][185]. SARS-CoV uses the same receptor as SARS-CoV-2 to infect cells, suggesting that both viruses may have similar life cycles [170]. However, the binding affinity of the S protein of SARS-CoV-2 to the human receptor is much higher than that of SARS-CoV [186]. This may be due to sequence differences, which makes the binding affinity of SARS-CoV-2 to ACE2 10 to 20 times higher than that of SARS-CoV, thus, enhancing the spread of SARS-CoV-2 [187][188]. The uncontrolled expansion of the virus is giving rise to new variants, with enhanced infectivity in some cases, that can challenge the control of the pandemic and compromise the efficiency of recently developed vaccines. Both, mutations in S protein leading to enhanced affinity for ACE2 receptor, and mutations reducing neutralizing activity of antibodies (immune escape), may be related to higher infectivity of new variants [189]. Since all SARS-CoV-2 variants use ACE2 for entry into the host cell, it is predicted that strategies targeting ACE2, should be effective in reducing infection by new variants. These strategies include BET inhibition, as explained below.
Once SARS-CoV-2 has entered the cell, the genetic material is released into the cytoplasm and starts translation. The first and only region directly translated from the genome is that coding for nsps (ORF1ab) [190]. Polyproteins 1ab and 1a are processed by the action of chymotrypsin-like protease (3CLpro, or main protease (Mpro)) and one or two papain-like proteases (PLpro) that are encoded by the virus [191]. The RTC locates in double membrane vesicles creating a protective microenvironment for replication of genomic RNA and transcription of subgenomic mRNAs. The subgenomic mRNAs are translated into accessory and structural proteins M, S and E that are isolated in the endoplasmic reticulum and then translocated to the endoplasmic reticulum-Golgi intermediate compartment. Subsequently they interact with the newly produced genomic RNA encapsidated by N protein, resulting in the formation of vesicles that are exported out of the cell through exocytosis [190][192][193].
3.2. SARS-CoV-2 Induced Immune Response and BET Proteins
The immune response is activated through recognition of both pathogen-associated and damage-associated molecular patterns by cell surface and intracellular pattern recognition receptors. In this scenario, toll-like family of pattern recognition receptors (TLR) play an important function. Expression of both TLR3 and TLR4 is upregulated by SARS-CoV-2 [194][195]. In addition, S protein interacts with and activates TLR4 [196][197]. BETs have been shown to positively regulate TLR4 expression in pancreatic ductal adenocarcinoma and in acute myocardial infarction rodent models [198][199]. In turn, TLR3 is activated by foreign RNAs molecules (particularly dsRNAs derived from virus replication), and it has been established that TLR3-induced acute airway inflammation and remodeling is efficiently neutralized by BET inhibitors, as it depends on BRD4 [200].
TLR3 and TLR4 stimulation results in NF-ĸB activation [161]. Of note, SARS-CoV-2 infection results in higher NF-ĸB pathway activation [201]. Notably, BRD4 control of innate immunity largely relies on BRD4 regulation of canonical NF-ĸB pathway, associated with transcription factor RELA. Under normal conditions, IĸBα blocks RELA in the cytoplasm. Inflammation-associated activation of IĸB kinases results in IĸBα phosphorylation, which is ubiquitinated and then targeted for degradation. Liberated RELA, undergoes translocation to the nucleus for regulation of inflammatory and immunomodulatory genes. RELA-mediated transcription activation depends on BRD4. Transcription activation by RELA requires its acetylation at K310, which is recognized by BRD4 [202]. BRD4 binding seems to stabilize RELA, since BET inhibition or BRD4 depletion leads to RELA ubiquitination and degradation [202][203]. In turn, RELA acetylation requires phosphorylation at S276 [204], which also depends on BRD4 [205]. Phosphorylation-coupled acetylation of RELA has been shown to facilitate BRD4 binding and recruitment of P-TEFb for transcriptional elongation of inflammatory cytokine genes upon RSV viral infection [204].
Activated NF-ĸB cooperates with Interferon Regulatory Factor (IRF) 3 to induce proinflammatory cytokines like type I interferon molecules (IFNs) and Tumor Necrosis Factor (TNF) [161]. Notably, upregulation of both types of molecules has been observed in COVID-19 patients [195]. IRFs are the main transcription factors involved in production of IFNs and are key regulators of antiviral immunity. It has been described that following RSV viral infection, the BRD4/RELA complex recruits the P-TEFb component CDK9 to IRF1 and IRF7 promoters for enhanced expression, and BRD4 inhibition has proven to alleviate viral-associated inflammation in this system [200]. Besides, it has been shown that virus infection in macrophages downregulates BRD3 expression and that BRD3 depletion impairs virus-mediated production of IFN-ß [206].
The tumorigenesis-associated JAK-STAT pathway also cooperates with NF-ĸB in triggering the immune response [207]. Through extracellular stimuli (mainly interleukin-6), the membrane receptor-associated Janus kinase (JAK) activates Signal Transducer and Activator of Transcription (STAT) factors to regulate the expression of cytokine-responsive genes [208]. JAK-STAT activation results in phosphorylated STAT, which enters the nucleus to activate transcription of IFN-stimulated genes. In human pluripotent stem cell-derived cardiac organoids (hPSC-COs), Mills et al. have reported that simulation of the COVID-19-associated CS leads to phosphorylation of STAT1 at S727 site [8]. On the other hand, BET inhibition has shown to efficiently inhibit the phosphorylation of STAT3 [209]. Moreover, combined inhibition of BET and JAK proteins has been shown to efficiently reverse inflammation linked to bone marrow fibrosis [210]. Brd4 siRNA delivery through liposome nanoparticles efficiently suppresses RELA and STAT3 activation in LPS-induced mouse models of inflammation [211].
Another important determinant of the immune response triggered by SARS-CoV-2 is NLRP3, which is well expressed in various cell types such as lung epithelial, kidney, cardiac, endothelial, hematopoietic and innate immune cells [212]. NLRP3 is the most studied component of the inflammosomes, which are multiprotein oligomers of the innate immune system responsible for the activation of the inflammatory response. Inflammosomes strikingly participate in Caspase 1 activation, which leads to the induction of pyroptosis, a newly introduced type of programmed cell death associated with inflammation [213]. It has been shown that BRD4 inhibition prevents proliferation and epithelial to mesenchymal transition in renal cell carcinoma by increasing NLRP3 levels, which results in activated Caspase 1 and pyroptosis [214].
Besides triggering inflammation, SARS-CoV-2 infection, like other respiratory viral infections, is linked to oxidative stress of the epithelium, whose cells activate the transcription factor Nuclear factor erythroid-derived 2-Related Factor 2 (NRF2) for protection against oxidation and inflammation [215][216]. Notably, it has been described that BRD4 downregulation or BET inhibitors lead to NRF2 stabilization, which results in decreased reactive oxygen species production [217].
TNF is a well-known mediator of inflammation-associated heart failure, which induces systolic dysfunction [218]. Indeed, simulated SARS-CoV-2 infection by TNF treatment of hPSC-COs also leads to systolic dysfunction [8]. Although quite different in origin, atherosclerosis and heart failure have in common the involvement of TNF in mediating the inflammatory response. It has been shown that TNF-mediated inflammation in endothelial cells directs the formation of RELA- and BRD4-dependent SEs, being BET inhibition able to abrogate SEs-derived transcription and atherosclerosis [219]. These types of SEs are regulated in a highly dynamic way. They are tightly associated with disease [36] and have been denominated “latent enhancers”, being usually formed in response to noxious stimuli in terminally differentiated cells [220]. In these conditions they are flooded with transcription-associated proteins, sharing many features with SEs. Once stimulus ceases, most of them remain in a latent state of memory, which enables faster and greater induction by next stimulation. Cardiac hypertrophy promotes great changes in methylation and acetylation of chromatin leading to activation of a great number of enhancers [221]. In heart failure models, BRD4 occupies the majority of activated enhancers and BET inhibition disturbs associated transcription, suppressing cardiomyocyte hypertrophy [222].
3.3. BET Inhibition for COVID-19 Treatment
Since organ damage associated with SARS-CoV-2 infection is tightly linked to overactivation of the immune response, a way to avoid damage is to neutralize hyperinflammation. On top of that, blocking virus recognition and entry into host cells will prevent triggering of the immune response, which will also result in aborted inflammation. BET inhibition has a lot to do with both approaches. We have highlighted the fundamental role BET proteins play in the control of the immune response, and we have indicated that expression of the main receptor in host cells for SARS-CoV-2 entry, the ACE2 protein, but also that of the associated TMPRSS2 protease, is under the control of BETs. Thus, BET inhibition, by interfering at different levels of the virus infection, may result in beneficial outcomes when used for treating COVID-19. It has been indicated that ACE2 expression is stimulated by activation of the immune response [223], and early in SARS-CoV-2 research became clear that antagonizing ACE2 and/or TMPRSS2 could be of interest for COVID-19 therapies [179][224][225][226][227].
From an unbiased CRISPRi screen to uncover druggable pathways controlling SARS-CoV-2 S protein binding to human cells, Tian et al. have determined that BRD2 is a key player of the cellular response to SARS-CoV-2 infection [10]. In this screen, they used Calu-3 cells, a lung epithelial cancer cell line that endogenously expresses ACE2. As expected, ACE2 downregulation was the major cause of impaired S protein binding. However, they also found BRD2 among downregulated genes leading to decreased S protein binding. In fact, they showed that downregulation of BRD2 correlated with lower levels of ACE2 transcript and thereby of protein. Overexpression of the full BRD2 protein recovered ACE2 transcriptional levels, demonstrating that BRD2 is required for ACE2 expression. Furthermore, it was shown that downregulation of BRD2 produced a complete inhibition of viral replication in these cells, showing levels similar to those observed when ACE2 was downregulated. The use of BET inhibitors produced effects similar to those observed upon BRD2 downregulation, with reduced ACE2 mRNA levels and S protein binding. Decreased ACE2 mRNA levels was observed in both primary human bronchial epithelial cells and cardiomyocytes. Viral replication was also affected by BET inhibitors, and in a similar way to that observed when downregulating BRD2 or ACE2. In addition, BET inhibition led to marked downregulation of genes that are involved in the response to type I IFN, whose expression is induced by SARS-CoV-2 both in patients and in cell cultures [10]. Therefore, these results suggest that BRD2 could be used as a therapeutic target for the treatment of COVID-19.
Moreover, SARS-CoV-2 infection is known to cause cardiac damage and dysfunction in 20–30% of hospitalized patients [228] and in the absence of infection well known inflammatory mediators such as TNF are associated with heart failure [218]. Mills et al. used hPSC-COs models, phosphoproteomic studies and single nuclei RNA sequencing to identify therapeutic targets and treatments for cardiac dysfunction. They studied the effects of several proinflammatory cytokines that are increased in COVID-19 patients and observed that they produced cardiac dysfunction, being TNF associated with systolic dysfunction and combination of IFN-γ, IL-1β and poly(I:C) with diastolic dysfunction, which is one of the most common dysfunction observed in COVID-19 patients [229]. The cardiac CS produced by IFN-γ, IL-1β and poly(I:C) induced 91 phosphosites, including one site on STAT1 and two sites on BRD4. Specific inhibitors exist for both proteins. However, while the different treatments used to inhibit STAT1 phosphorylation did not prevent CS-induced diastolic dysfunction, of several BET inhibitors tested (INCB054329, JQ1, RXV-2157, apabetalone and ABBV-744), the first four showed protection. This study demonstrated that CS-mediated diastolic dysfunction is mediated by BRD4-dependent mechanisms that can be blocked using BET inhibitors. Also in mice, they showed that response in the heart triggered by SARS-CoV-2 infection was partially blocked by treatment with INCB054329. Pre-incubation of hPSC-COs with INCB054329 prior to infection reduced ACE2 expression and decreased intracellular viral RNA, demonstrating the potential of BET protein inhibitors to block SARS-CoV-2 infection and prevent dysfunction [8]. BET inhibitors with dual BDI and BDII activities may display side effects [43], making necessary to determine the selectivity of the inhibition. Importantly, molecules specifically inhibiting BDII, like RXV-2157 and apabetalone, efficiently blocked SARS-CoV-2 infection by decreasing ACE2 expression and thereby SARS-CoV-2 S protein binding, showing that selective inhibitors against BDII are potential candidates to prevent heart damage caused by COVID-19 [8].
In line with this, a more recent study carried out by Gilham et al., shows that apabetalone, as JQ1, produces downregulation of ACE2 expression in different cell types: Calu-3 cells, Vero E6 cell (monkey kidney epithelial cells), hepatocarcinoma cells HepG2 and Huh-7, and primary human hepatocytes [7]. Decrease in mRNA levels is accompanied by a decrease in protein levels in Calu-3 and Vero E6 cells. Apabetalone treatment also decreased DPP4 expression in Calu-3 cells. DDP4 encodes for Dipeptidyl peptidase 4 (CD26), a potential cofactor for SARS-CoV-2 entry into the host cell, as its presence on the cell surface facilitates viral binding [230][231]. Apabetalone and other BET protein inhibitors attenuate SARS-CoV-2 S protein binding and abrogate SARS-CoV-2 infection. These results and its well-established safety profile, together with the dual mechanism of action simultaneously combating hyperinflammation [232][233][234][235] and ACE2-mediated viral entry, make apabetalone a good candidate for treating SARS-CoV-2 infection [7]. Indeed, a clinical trial with apabetalone for COVID-19 treatment has been approved: NCT04894266 identifier (https://clinicaltrials.gov/, accessed on 22 July 2021).
Additional studies have reinforced the interest on BET targeting as an effective tool against SARS-CoV-2 infection. Expression of the key host proteins ACE2 and TMPRSS2 mediating virus entry in the cell is regulated by androgens [9][236] and transcriptional repression of the androgen receptor (AR) enhanceosome with AR or BET inhibitors suppressing SARS-CoV-2 infection in vitro [9]. Studies have shown that AR, ACE2 and TMPRSS2 are co-expressed in various types of human and murine lung epithelial cells, including alveolar and bronchial cells. Adult immune-competent C57BL/6 male mice were castrated to create an androgen-deprived condition and compared with non-castrated mice and with castrated mice treated for 5 days with testosterone. The experiments showed that Tmprss2 and Ace2 are positively regulated by androgens [9]. In the AR-positive LNCaP prostate cancer cells, which can be infected by SARS-CoV-2, it has been shown that blocking AR signaling with the use of different AR antagonists approved for prostate cancer treatment affects the infective capacity of SARS-CoV-2, showing a dose-dependent decrease in the expression of TMPRSS2 and ACE2. Likewise, the use of different BET protein inhibitors showed decreased expression of TMPRSS2 and ACE2, suggesting once again that BET proteins play a role in regulating the expression of the SARS-CoV-2 entry factor ACE2 and that this is independent of AR regulation, supporting the idea that inhibition of BET proteins may be useful to mitigate SARS-CoV-2 infection [9].
SUPT16H protein together with SSRP1 constitute FACT (FAcilitates Chromatin Transcription), a heterodimeric histone chaperone associated with chromatin remodeling during gene transcription [237]. BRD4 stabilizes SUPT16H by recognizing SUPT16H acetylation at K647 [238]. Targeting SUPT16H by RNAi-based approaches or pharmacological inhibition leads to the induction of interferons and interferon-stimulated genes, efficiently inhibiting SARS-CoV-2 infection, but also infection by other viruses like Zika and influenza [238]. This raises the question whether the effects of BET inhibitors on SARS-CoV-2 infection are mediated, at least in part, by altered SUPT16H stability. Besides BET proteins, another critical druggable target identified in SARS-CoV-2 interactome is mTOR [6]. It has been shown that the dual BET/mTOR-PI3K-α SF2523 inhibitor effectively blocks SARS-CoV-2 replication in lung bronchial epithelial cells in vitro [239]. Moreover, synergistic effects are observed when SF2523 is combined with remdesivir. Additional molecules like flavonoids and allergen fragrance molecules have been proposed as compounds of interest to interfere with SARS-CoV-2 infection through BET inhibition [240][241].
In sum, BET inhibition for beneficial effects on SARS-CoV-2 infection seems to operate at different levels (Figure 3). By targeting BETs, different processes/pathways relevant for virus infection can be simultaneously interfered. As explained, BET inhibition should result in attenuated inflammation but also in decreased ACE2 expression, which will result in hampered infection, collectively leading to reduced tissue damage. Besides, direct targeting of host proteins by viral proteins may also have consequences but it is an additional piece of the landscape at best. In the case of E protein interacting with BETs we do not know at present whether interaction leads to significant impaired BET function. This seems not to be the case, as Tian et al. have reported that E protein overexpression has mild and non-overlapping effects on transcriptome in comparison with the effect of BRD2 knockdown or BET inhibition [10]. In the case that the bromodomains mediate the interaction with E protein we can anticipate that the use of BET inhibitors may be detrimental for the cell, as inhibitors can enhance E protein-mediated interference at bromodomain level. However, we can speculate about the use of well-defined concentrations of BET inhibitors resulting in E protein dissociation without grossly affecting chromatin attachment of BET proteins, thus resulting in a benefit for the cell. Nevertheless, as demonstrated, the use of BET inhibitors has proven to be an efficient tool in fighting SARS-CoV-2 infection effects for many other reasons.
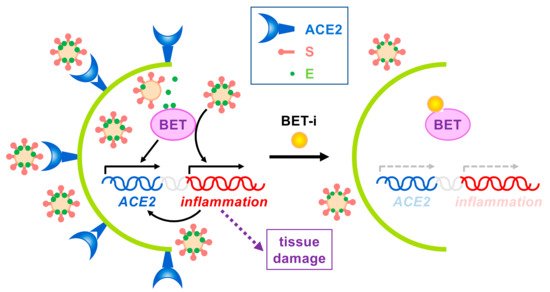
Figure 3. Effects of BET inhibition on SARS-CoV-2 infection. On one hand, expression of the SARS-CoV-2 receptor ACE2 depends on BET proteins. On the other hand, virus infection triggers the immune response, leading to BET-dependent activation of inflammation, which in turn may also activate ACE2 expression. Uncontrolled inflammation may cause severe tissue damage. Besides, SARS-CoV-2 E protein interacts with BETs, but no associated effects have been reported to date. The use of BET inhibitors (BET-i) attenuates ACE2 expression and counteracts inflammation, thus, reducing infection and tissue damage.
References
- Alekseyenko, A.A.; Walsh, E.M.; Wang, X.; Grayson, A.R.; Hsi, P.T.; Kharchenko, P.V.; Kuroda, M.I.; French, C.A. The oncogenic BRD4-NUT chromatin regulator drives aberrant transcription within large topological domains. Genes Dev. 2015, 29, 1507–1523.
- French, C.A.; Miyoshi, I.; Kubonishi, I.; Grier, H.E.; Perez-Atayde, A.R.; Fletcher, J.A. BRD4-NUT fusion oncogene: A novel mechanism in aggressive carcinoma. Cancer Res. 2003, 63, 304–307.
- Gaucher, J.; Boussouar, F.; Montellier, E.; Curtet, S.; Buchou, T.; Bertrand, S.; Hery, P.; Jounier, S.; Depaux, A.; Vitte, A.L.; et al. Bromodomain-dependent stage-specific male genome programming by Brdt. EMBO J. 2012, 31, 3809–3820.
- LeRoy, G.; Chepelev, I.; Di Maggio, P.A.; Blanco, M.A.; Zee, B.M.; Zhao, K.; Garcia, B.A. Proteogenomic characterization and mapping of nucleosomes decoded by Brd and HP1 proteins. Genome Biol. 2012, 13, R68.
- Shang, E.; Salazar, G.; Crowley, T.E.; Wang, X.; Lopez, R.A.; Wang, X.; Wolgemuth, D.J. Identification of unique, differentiation stage-specific patterns of expression of the bromodomain-containing genes Brd2, Brd3, Brd4, and Brdt in the mouse testis. Gene Expr. Patterns 2004, 4, 513–519.
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468.
- Gilham, D.; Smith, A.L.; Fu, L.; Moore, D.Y.; Muralidharan, A.; Reid, S.P.M.; Stotz, S.C.; Johansson, J.O.; Sweeney, M.; Wong, N.C.W.; et al. Bromodomain and extraterminal protein inhibitor, apabetalone (RVX-208), reduces ACE2 expression and attenuates SARS-CoV-2 infection in vitro. Biomedicines 2021, 9, 437.
- Mills, R.J.; Humphrey, S.J.; Fortuna, P.R.J.; Lor, M.; Foster, S.R.; Quaife-Ryan, G.A.; Johnston, R.L.; Dumenil, T.; Bishop, C.; Rudraraju, R.; et al. BET inhibition blocks inflammation-induced cardiac dysfunction and SARS-CoV-2 infection. Cell 2021, 184, 2167–2182.
- Qiao, Y.; Wang, X.M.; Mannan, R.; Pitchiaya, S.; Zhang, Y.; Wotring, J.W.; Xiao, L.; Robinson, D.R.; Wu, Y.M.; Tien, J.C.; et al. Targeting transcriptional regulation of SARS-CoV-2 entry factors ACE2 and TMPRSS2. Proc. Natl. Acad. Sci. USA 2020.
- Tian, R.; Samelson, A.J.; Rezelj, V.V.; Chen, M.; Ramadoss, G.N.; Guo, X.; Kain, A.M.; Tran, Q.D.; Lim, S.A.; Lui, I.; et al. BRD2 inhibition blocks SARS-CoV-2 infection in vitro by reducing transcription of the host cell receptor ACE2. bioRxiv 2021.
- You, J.; Croyle, J.L.; Nishimura, A.; Ozato, K.; Howley, P.M. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell 2004, 117, 349–360.
- McBride, A.A.; Jang, M.K. Current understanding of the role of the Brd4 protein in the papillomavirus lifecycle. Viruses 2013, 5, 1374.
- Wu, S.Y.; Nin, D.S.; Lee, A.Y.; Simanski, S.; Kodadek, T.; Chiang, C.M. BRD4 phosphorylation regulates HPV E2-mediated viral transcription, origin replication, and cellular MMP-9 expression. Cell Rep. 2016, 16, 1733–1748.
- Platt, G.M.; Simpson, G.R.; Mittnacht, S.; Schulz, T.F. Latent nuclear antigen of Kaposi’s sarcoma-associated herpesvirus interacts with RING3, a homolog of the Drosophila female sterile homeotic (fsh) gene. J. Virol. 1999, 73, 9789–9795.
- Ottinger, M.; Christalla, T.; Nathan, K.; Brinkmann, M.M.; Viejo-Borbolla, A.; Schulz, T.F. Kaposi’s sarcoma-associated herpesvirus LANA-1 interacts with the short variant of BRD4 and releases cells from a BRD4- and BRD2/RING3-induced G1 cell cycle arrest. J. Virol. 2006, 80, 10772–10786.
- Viejo-Borbolla, A.; Ottinger, M.; Bruning, E.; Burger, A.; Konig, R.; Kati, E.; Sheldon, J.A.; Schulz, T.F. Brd2/RING3 interacts with a chromatin-binding domain in the Kaposi’s Sarcoma-associated herpesvirus latency-associated nuclear antigen 1 (LANA-1) that is required for multiple functions of LANA-1. J. Virol. 2005, 79, 13618–13629.
- You, J.; Srinivasan, V.; Denis, G.V.; Harrington, W.J., Jr.; Ballestas, M.E.; Kaye, K.M.; Howley, P.M. Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen interacts with bromodomain protein Brd4 on host mitotic chromosomes. J. Virol. 2006, 80, 8909–8919.
- Lotke, R.; Schneeweiss, U.; Pietrek, M.; Gunther, T.; Grundhoff, A.; Weidner-Glunde, M.; Schulz, T.F. Brd/BET proteins influence the genome-wide localization of the Kaposi’s Sarcoma-associated herpesvirus and murine gammaherpesvirus major latency proteins. Front. Microbiol. 2020, 11, 591778.
- Ottinger, M.; Pliquet, D.; Christalla, T.; Frank, R.; Stewart, J.P.; Schulz, T.F. The interaction of the gammaherpesvirus 68 orf73 protein with cellular BET proteins affects the activation of cell cycle promoters. J. Virol. 2009, 83, 4423–4434.
- Lin, A.; Wang, S.; Nguyen, T.; Shire, K.; Frappier, L. The EBNA1 protein of Epstein-Barr virus functionally interacts with Brd4. J. Virol. 2008, 82, 12009–12019.
- Keck, K.M.; Moquin, S.A.; He, A.; Fernandez, S.G.; Somberg, J.J.; Liu, S.M.; Martinez, D.M.; Miranda, J.L. Bromodomain and extraterminal inhibitors block the Epstein-Barr virus lytic cycle at two distinct steps. J. Biol Chem. 2017, 292, 13284–13295.
- Aiyer, S.; Swapna, G.V.; Malani, N.; Aramini, J.M.; Schneider, W.M.; Plumb, M.R.; Ghanem, M.; Larue, R.C.; Sharma, A.; Studamire, B.; et al. Altering murine leukemia virus integration through disruption of the integrase and BET protein family interaction. Nucleic Acids Res. 2014, 42, 5917–5928.
- De Rijck, J.; de Kogel, C.; Demeulemeester, J.; Vets, S.; El Ashkar, S.; Malani, N.; Bushman, F.D.; Landuyt, B.; Husson, S.J.; Busschots, K.; et al. The BET family of proteins targets moloney murine leukemia virus integration near transcription start sites. Cell Rep. 2013, 5, 886–894.
- Gupta, S.S.; Maetzig, T.; Maertens, G.N.; Sharif, A.; Rothe, M.; Weidner-Glunde, M.; Galla, M.; Schambach, A.; Cherepanov, P.; Schulz, T.F. Bromo- and extraterminal domain chromatin regulators serve as cofactors for murine leukemia virus integration. J. Virol. 2013, 87, 12721–12736.
- Sharma, A.; Larue, R.C.; Plumb, M.R.; Malani, N.; Male, F.; Slaughter, A.; Kessl, J.J.; Shkriabai, N.; Coward, E.; Aiyer, S.S.; et al. BET proteins promote efficient murine leukemia virus integration at transcription start sites. Proc. Natl. Acad. Sci. USA 2013, 110, 12036–12041.
- Gallay, K.; Blot, G.; Chahpazoff, M.; Yajjou-Hamalian, H.; Confort, M.P.; De Boisseson, C.; Leroux, A.; Luengo, C.; Fiorini, F.; Lavigne, M.; et al. In vitro, in cellulo and structural characterizations of the interaction between the integrase of Porcine Endogenous Retrovirus A/C and proteins of the BET family. Virology 2019, 532, 69–81.
- Bisgrove, D.A.; Mahmoudi, T.; Henklein, P.; Verdin, E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 13690–13695.
- Li, Z.; Guo, J.; Wu, Y.; Zhou, Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013, 41, 277–287.
- Zhu, J.; Gaiha, G.D.; John, S.P.; Pertel, T.; Chin, C.R.; Gao, G.; Qu, H.; Walker, B.D.; Elledge, S.J.; Brass, A.L. Reactivation of latent HIV-1 by inhibition of BRD4. Cell Rep. 2012, 2, 807–816.
- Li, Z.; Mbonye, U.; Feng, Z.; Wang, X.; Gao, X.; Karn, J.; Zhou, Q. The KAT5-Acetyl-Histone4-Brd4 axis silences HIV-1 transcription and promotes viral latency. PLoS Pathog. 2018, 14, e1007012.
- Cho, W.K.; Zhou, M.; Jang, M.K.; Huang, K.; Jeong, S.J.; Ozato, K.; Brady, J.N. Modulation of the Brd4/P-TEFb interaction by the human T-lymphotropic virus type 1 tax protein. J. Virol. 2007, 81, 11179–11186.
- Groves, I.J.; Jackson, S.E.; Poole, E.L.; Nachshon, A.; Rozman, B.; Schwartz, M.; Prinjha, R.K.; Tough, D.F.; Sinclair, J.H.; Wills, M.R. Bromodomain proteins regulate human cytomegalovirus latency and reactivation allowing epigenetic therapeutic intervention. Proc. Natl. Acad. Sci. USA 2021, 118.
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer 2021, 124, 1478–1490.
- Bechter, O.; Schöffski, P. Make your best BET: The emerging role of BET inhibitor treatment in malignant tumors. Pharmacol. Ther. 2020, 208, 107479.
- Andrieu, G.P.; Shafran, J.S.; Deeney, J.T.; Bharadwaj, K.R.; Rangarajan, A.; Denis, G.V. BET proteins in abnormal metabolism, inflammation, and the breast cancer microenvironment. J. Leukoc. Biol. 2018, 104, 265–274.
- Kulikowski, E.; Rakai, B.D.; Wong, N.C.W. Inhibitors of bromodomain and extra-terminal proteins for treating multiple human diseases. Med. Res. Rev. 2021, 41, 223–245.
- Singh, M.B.; Sartor, G.C. BET bromodomains as novel epigenetic targets for brain health and disease. Neuropharmacology 2020, 181, 108306.
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533.
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917.
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073.
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J., 3rd. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674.
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528.
- Gilan, O.; Rioja, I.; Knezevic, K.; Bell, M.J.; Yeung, M.M.; Harker, N.R.; Lam, E.Y.N.; Chung, C.W.; Bamborough, P.; Petretich, M.; et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science 2020, 368, 387–394.
- Sun, Y.; Han, J.; Wang, Z.; Li, X.; Sun, Y.; Hu, Z. Safety and efficacy of bromodomain and extra-terminal inhibitors for the treatment of hematological malignancies and solid tumors: A systematic study of clinical trials. Front. Pharmacol. 2020, 11, 621093.
- Zaware, N.; Zhou, M.M. Bromodomain biology and drug discovery. Nat. Struct. Mol. Biol. 2019, 26, 870–879.
- Weidner-Glunde, M.; Ottinger, M.; Schulz, T.F. WHAT do viruses BET on? Front. Biosci. 2010, 15, 537–549.
- Belkina, A.C.; Denis, G.V. BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 2012, 12, 465–477.
- Wang, N.; Wu, R.; Tang, D.; Kang, R. The BET family in immunity and disease. Signal. Transduct. Target Ther. 2021, 6, 23.
- Paillisson, A.; Levasseur, A.; Gouret, P.; Callebaut, I.; Bontoux, M.; Pontarotti, P.; Monget, P. Bromodomain testis-specific protein is expressed in mouse oocyte and evolves faster than its ubiquitously expressed paralogs BRD2, -3, and -4. Genomics 2007, 89, 215–223.
- Wu, S.Y.; Chiang, C.M. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J. Biol. Chem. 2007, 282, 13141–13145.
- Zeng, L.; Zhou, M.M. Bromodomain: An acetyl-lysine binding domain. FEBS Lett. 2002, 513, 124–128.
- Gamsjaeger, R.; Webb, S.R.; Lamonica, J.M.; Billin, A.; Blobel, G.A.; Mackay, J.P. Structural basis and specificity of acetylated transcription factor GATA1 recognition by BET family bromodomain protein Brd3. Mol. Cell Biol. 2011, 31, 2632–2640.
- Lamonica, J.M.; Deng, W.; Kadauke, S.; Campbell, A.E.; Gamsjaeger, R.; Wang, H.; Cheng, Y.; Billin, A.N.; Hardison, R.C.; Mackay, J.P.; et al. Bromodomain protein Brd3 associates with acetylated GATA1 to promote its chromatin occupancy at erythroid target genes. Proc. Natl. Acad. Sci. USA 2011, 108, E159–E168.
- Stonestrom, A.J.; Hsu, S.C.; Jahn, K.S.; Huang, P.; Keller, C.A.; Giardine, B.M.; Kadauke, S.; Campbell, A.E.; Evans, P.; Hardison, R.C.; et al. Functions of BET proteins in erythroid gene expression. Blood 2015, 125, 2825–2834.
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.S.; Brady, J.N.; Ozato, K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 2005, 19, 523–534.
- Dey, A.; Chitsaz, F.; Abbasi, A.; Misteli, T.; Ozato, K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. USA 2003, 100, 8758–8763.
- Ito, T.; Umehara, T.; Sasaki, K.; Nakamura, Y.; Nishino, N.; Terada, T.; Shirouzu, M.; Padmanabhan, B.; Yokoyama, S.; Ito, A.; et al. Real-time imaging of histone H4K12-specific acetylation determines the modes of action of histone deacetylase and bromodomain inhibitors. Chem. Biol. 2011, 18, 495–507.
- Kanno, T.; Kanno, Y.; Siegel, R.M.; Jang, M.K.; Lenardo, M.J.; Ozato, K. Selective recognition of acetylated histones by bromodomain proteins visualized in living cells. Mol. Cell 2004, 13, 33–43.
- LeRoy, G.; Rickards, B.; Flint, S.J. The double bromodomain proteins Brd2 and Brd3 couple histone acetylation to transcription. Mol. Cell 2008, 30, 51–60.
- Morinière, J.; Rousseaux, S.; Steuerwald, U.; Soler-Lopez, M.; Curtet, S.; Vitte, A.L.; Govin, J.; Gaucher, J.; Sadoul, K.; Hart, D.J.; et al. Cooperative binding of two acetylation marks on a histone tail by a single bromodomain. Nature 2009, 461, 664–668.
- Sasaki, K.; Ito, T.; Nishino, N.; Khochbin, S.; Yoshida, M. Real-time imaging of histone H4 hyperacetylation in living cells. Proc. Natl. Acad. Sci. USA 2009, 106, 16257–16262.
- Umehara, T.; Nakamura, Y.; Jang, M.K.; Nakano, K.; Tanaka, A.; Ozato, K.; Padmanabhan, B.; Yokoyama, S. Structural basis for acetylated histone H4 recognition by the human BRD2 bromodomain. J. Biol. Chem. 2010, 285, 7610–7618.
- Umehara, T.; Nakamura, Y.; Wakamori, M.; Ozato, K.; Yokoyama, S.; Padmanabhan, B. Structural implications for K5/K12-di-acetylated histone H4 recognition by the second bromodomain of BRD2. FEBS Lett. 2010, 584, 3901–3908.
- García-Gutiérrez, P.; Mundi, M.; García-Domínguez, M. Association of bromodomain BET proteins with chromatin requires dimerization through the conserved motif B. J. Cell Sci. 2012, 125, 3671–3680.
- García-Gutiérrez, P.; Juárez-Vicente, F.; Wolgemuth, D.J.; García-Domínguez, M. Pleiotrophin antagonizes Brd2 during neuronal differentiation. J. Cell Sci. 2014, 127, 2554–2564.
- Luna-Peláez, N.; García-Domínguez, M. Lyar-mediated recruitment of Brd2 to the chromatin attenuates nanog downregulation following induction of differentiation. J. Mol. Biol. 2018, 430, 1084–1097.
- Luna-Peláez, N.; March-Díaz, R.; Ceballos-Chávez, M.; Guerrero-Martínez, J.A.; Grazioli, P.; García-Gutiérrez, P.; Vaccari, T.; Massa, V.; Reyes, J.C.; García-Domínguez, M. The Cornelia de Lange Syndrome-associated factor NIPBL interacts with BRD4 ET domain for transcription control of a common set of genes. Cell Death Dis. 2019, 10, 548.
- Rahman, S.; Sowa, M.E.; Ottinger, M.; Smith, J.A.; Shi, Y.; Harper, J.W.; Howley, P.M. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell Biol. 2011, 31, 2641–2652.
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545.
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 2020, 181, 1036–1045.e1039.
- Denis, G.V.; McComb, M.E.; Faller, D.V.; Sinha, A.; Romesser, P.B.; Costello, C.E. Identification of transcription complexes that contain the double bromodomain protein Brd2 and chromatin remodeling machines. J. Proteome Res. 2006, 5, 502–511.
- Denis, G.V.; Vaziri, C.; Guo, N.; Faller, D.V. RING3 kinase transactivates promoters of cell cycle regulatory genes through E2F. Cell Growth Differ. 2000, 11, 417–424.
- Sinha, A.; Faller, D.V.; Denis, G.V. Bromodomain analysis of Brd2-dependent transcriptional activation of cyclin A. Biochem. J. 2005, 387, 257–269.
- Dey, A.; Ellenberg, J.; Farina, A.; Coleman, A.E.; Maruyama, T.; Sciortino, S.; Lippincott-Schwartz, J.; Ozato, K. A bromodomain protein, MCAP, associates with mitotic chromosomes and affects G(2)-to-M transition. Mol. Cell Biol. 2000, 20, 6537–6549.
- Mochizuki, K.; Nishiyama, A.; Jang, M.K.; Dey, A.; Ghosh, A.; Tamura, T.; Natsume, H.; Yao, H.; Ozato, K. The bromodomain protein Brd4 stimulates G1 gene transcription and promotes progression to S phase. J. Biol. Chem. 2008, 283, 9040–9048.
- Yang, Z.; He, N.; Zhou, Q. Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol. Cell Biol. 2008, 28, 967–976.
- Liu, D.; Matzuk, M.M.; Sung, W.K.; Guo, Q.; Wang, P.; Wolgemuth, D.J. Cyclin A1 is required for meiosis in the male mouse. Nat. Genet. 1998, 20, 377–380.
- Shang, E.; Nickerson, H.D.; Wen, D.; Wang, X.; Wolgemuth, D.J. The first bromodomain of Brdt, a testis-specific member of the BET sub-family of double-bromodomain-containing proteins, is essential for male germ cell differentiation. Development 2007, 134, 3507–3515.
- Shen, Z.; Zhang, G.; Yang, Y.; Li, M.; Yang, S.; Peng, G. Lysine 164 is critical for SARS-CoV-2 Nsp1 inhibition of host gene expression. J. Gen. Virol. 2021, 102.
- Zhao, R.; Nakamura, T.; Fu, Y.; Lazar, Z.; Spector, D.L. Gene bookmarking accelerates the kinetics of post-mitotic transcriptional re-activation. Nat. Cell Biol. 2011, 13, 1295–1304.
- Pivot-Pajot, C.; Caron, C.; Govin, J.; Vion, A.; Rousseaux, S.; Khochbin, S. Acetylation-dependent chromatin reorganization by BRDT, a testis-specific bromodomain-containing protein. Mol. Cell Biol. 2003, 23, 5354–5365.
- Wang, R.; Li, Q.; Helfer, C.M.; Jiao, J.; You, J. Bromodomain protein Brd4 associated with acetylated chromatin is important for maintenance of higher-order chromatin structure. J. Biol. Chem. 2012, 287, 10738–10752.
- Hsu, S.C.; Gilgenast, T.G.; Bartman, C.R.; Edwards, C.R.; Stonestrom, A.J.; Huang, P.; Emerson, D.J.; Evans, P.; Werner, M.T.; Keller, C.A.; et al. The BET protein BRD2 cooperates with CTCF to enforce transcriptional and architectural boundaries. Mol. Cell 2017, 66, 102–116.
- Itzen, F.; Greifenberg, A.K.; Bosken, C.A.; Geyer, M. Brd4 activates P-TEFb for RNA polymerase II CTD phosphorylation. Nucleic Acids Res. 2014, 42, 7577–7590.
- Devaiah, B.N.; Lewis, B.A.; Cherman, N.; Hewitt, M.C.; Albrecht, B.K.; Robey, P.G.; Ozato, K.; Sims, R.J., 3rd; Singer, D.S. BRD4 is an atypical kinase that phosphorylates serine2 of the RNA polymerase II carboxy-terminal domain. Proc. Natl. Acad. Sci. USA 2012, 109, 6927–6932.
- Wada, T.; Takagi, T.; Yamaguchi, Y.; Watanabe, D.; Handa, H. Evidence that P-TEFb alleviates the negative effect of DSIF on RNA polymerase II-dependent transcription in vitro. EMBO J. 1998, 17, 7395–7403.
- Yamada, T.; Yamaguchi, Y.; Inukai, N.; Okamoto, S.; Mura, T.; Handa, H. P-TEFb-mediated phosphorylation of hSpt5 C-terminal repeats is critical for processive transcription elongation. Mol. Cell 2006, 21, 227–237.
- Yamaguchi, Y.; Takagi, T.; Wada, T.; Yano, K.; Furuya, A.; Sugimoto, S.; Hasegawa, J.; Handa, H. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell 1999, 97, 41–51.
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-Andre, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947.
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319.
- Sengupta, S.; George, R.E. Super-enhancer-driven transcriptional dependencies in cancer. Trends Cancer 2017, 3, 269–281.
- Shi, J.; Vakoc, C.R. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 2014, 54, 728–736.
- Gyuris, A.; Donovan, D.J.; Seymour, K.A.; Lovasco, L.A.; Smilowitz, N.R.; Halperin, A.L.; Klysik, J.E.; Freiman, R.N. The chromatin-targeting protein Brd2 is required for neural tube closure and embryogenesis. Biochim. Biophys. Acta 2009, 1789, 413–421.
- Houzelstein, D.; Bullock, S.L.; Lynch, D.E.; Grigorieva, E.F.; Wilson, V.A.; Beddington, R.S. Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein Brd4. Mol. Cell Biol. 2002, 22, 3794–3802.
- Shang, E.; Wang, X.; Wen, D.; Greenberg, D.A.; Wolgemuth, D.J. Double bromodomain-containing gene Brd2 is essential for embryonic development in mouse. Dev. Dyn. 2009, 238, 908–917.
- Wang, F.; Liu, H.; Blanton, W.P.; Belkina, A.; Lebrasseur, N.K.; Denis, G.V. Brd2 disruption in mice causes severe obesity without Type 2 diabetes. Biochem. J. 2009, 425, 71–83.
- Velisek, L.; Shang, E.; Veliskova, J.; Chachua, T.; Macchiarulo, S.; Maglakelidze, G.; Wolgemuth, D.J.; Greenberg, D.A. GABAergic neuron deficit as an idiopathic generalized epilepsy mechanism: The role of BRD2 haploinsufficiency in juvenile myoclonic epilepsy. PLoS ONE 2011, 6, e23656.
- Korb, E.; Herre, M.; Zucker-Scharff, I.; Darnell, R.B.; Allis, C.D. BET protein Brd4 activates transcription in neurons and BET inhibitor Jq1 blocks memory in mice. Nat. Neurosci. 2015, 18, 1464–1473.
- McBride, A.A. The papillomavirus E2 proteins. Virology 2013, 445, 57–79.
- Rapp, B.; Pawellek, A.; Kraetzer, F.; Schaefer, M.; May, C.; Purdie, K.; Grassmann, K.; Iftner, T. Cell-type-specific separate regulation of the E6 and E7 promoters of human papillomavirus type 6a by the viral transcription factor E2. J. Virol. 1997, 71, 6956–6966.
- Bastien, N.; McBride, A.A. Interaction of the papillomavirus E2 protein with mitotic chromosomes. Virology 2000, 270, 124–134.
- McPhillips, M.G.; Oliveira, J.G.; Spindler, J.E.; Mitra, R.; McBride, A.A. Brd4 is required for e2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J. Virol. 2006, 80, 9530–9543.
- Ilves, I.; Maemets, K.; Silla, T.; Janikson, K.; Ustav, M. Brd4 is involved in multiple processes of the bovine papillomavirus type 1 life cycle. J. Virol. 2006, 80, 3660–3665.
- Lee, A.Y.; Chiang, C.M. Chromatin adaptor Brd4 modulates E2 transcription activity and protein stability. J. Biol. Chem. 2009, 284, 2778–2786.
- Schweiger, M.R.; You, J.; Howley, P.M. Bromodomain protein 4 mediates the papillomavirus E2 transcriptional activation function. J. Virol. 2006, 80, 4276–4285.
- Senechal, H.; Poirier, G.G.; Coulombe, B.; Laimins, L.A.; Archambault, J. Amino acid substitutions that specifically impair the transcriptional activity of papillomavirus E2 affect binding to the long isoform of Brd4. Virology 2007, 358, 10–17.
- Smith, J.A.; White, E.A.; Sowa, M.E.; Powell, M.L.; Ottinger, M.; Harper, J.W.; Howley, P.M. Genome-wide siRNA screen identifies SMCX, EP400, and Brd4 as E2-dependent regulators of human papillomavirus oncogene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 3752–3757.
- Wu, S.Y.; Lee, A.Y.; Hou, S.Y.; Kemper, J.K.; Erdjument-Bromage, H.; Tempst, P.; Chiang, C.M. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 2006, 20, 2383–2396.
- Wang, X.; Helfer, C.M.; Pancholi, N.; Bradner, J.E.; You, J. Recruitment of Brd4 to the human papillomavirus type 16 DNA replication complex is essential for replication of viral DNA. J. Virol. 2013, 87, 3871–3884.
- Sakakibara, N.; Chen, D.; Jang, M.K.; Kang, D.W.; Luecke, H.F.; Wu, S.Y.; Chiang, C.M.; McBride, A.A. Brd4 is displaced from HPV replication factories as they expand and amplify viral DNA. PLoS Pathog. 2013, 9, e1003777.
- Iftner, T.; Haedicke-Jarboui, J.; Wu, S.Y.; Chiang, C.M. Involvement of Brd4 in different steps of the papillomavirus life cycle. Virus Res. 2017, 231, 76–82.
- Bellanger, S.; Blachon, S.; Mechali, F.; Bonne-Andrea, C.; Thierry, F. High-risk but not low-risk HPV E2 proteins bind to the APC activators Cdh1 and Cdc20 and cause genomic instability. Cell Cycle 2005, 4, 1608–1615.
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191.
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869.
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; d’Agay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L.; et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 1995, 86, 1276–1280.
- Garber, A.C.; Hu, J.; Renne, R. Latency-associated nuclear antigen (LANA) cooperatively binds to two sites within the terminal repeat, and both sites contribute to the ability of LANA to suppress transcription and to facilitate DNA replication. J. Biol. Chem. 2002, 277, 27401–27411.
- Garber, A.C.; Shu, M.A.; Hu, J.; Renne, R. DNA binding and modulation of gene expression by the latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2001, 75, 7882–7892.
- Grundhoff, A.; Ganem, D. The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus permits replication of terminal repeat-containing plasmids. J. Virol. 2003, 77, 2779–2783.
- Hu, J.; Garber, A.C.; Renne, R. The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus supports latent DNA replication in dividing cells. J. Virol. 2002, 76, 11677–11687.
- Hu, J.; Renne, R. Characterization of the minimal replicator of Kaposi’s sarcoma-associated herpesvirus latent origin. J. Virol. 2005, 79, 2637–2642.
- Barbera, A.J.; Chodaparambil, J.V.; Kelley-Clarke, B.; Joukov, V.; Walter, J.C.; Luger, K.; Kaye, K.M. The nucleosomal surface as a docking station for Kaposi’s sarcoma herpesvirus LANA. Science 2006, 311, 856–861.
- Kelley-Clarke, B.; Ballestas, M.E.; Srinivasan, V.; Barbera, A.J.; Komatsu, T.; Harris, T.A.; Kazanjian, M.; Kaye, K.M. Determination of Kaposi’s sarcoma-associated herpesvirus C-terminal latency-associated nuclear antigen residues mediating chromosome association and DNA binding. J. Virol. 2007, 81, 4348–4356.
- Lim, C.; Lee, D.; Seo, T.; Choi, C.; Choe, J. Latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus functionally interacts with heterochromatin protein 1. J. Biol. Chem. 2003, 278, 7397–7405.
- Piolot, T.; Tramier, M.; Coppey, M.; Nicolas, J.C.; Marechal, V. Close but distinct regions of human herpesvirus 8 latency-associated nuclear antigen 1 are responsible for nuclear targeting and binding to human mitotic chromosomes. J. Virol. 2001, 75, 3948–3959.
- Sakakibara, S.; Ueda, K.; Nishimura, K.; Do, E.; Ohsaki, E.; Okuno, T.; Yamanishi, K. Accumulation of heterochromatin components on the terminal repeat sequence of Kaposi’s sarcoma-associated herpesvirus mediated by the latency-associated nuclear antigen. J. Virol. 2004, 78, 7299–7310.
- Shinohara, H.; Fukushi, M.; Higuchi, M.; Oie, M.; Hoshi, O.; Ushiki, T.; Hayashi, J.; Fujii, M. Chromosome binding site of latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus is essential for persistent episome maintenance and is functionally replaced by histone H1. J. Virol. 2002, 76, 12917–12924.
- Skalsky, R.L.; Hu, J.; Renne, R. Analysis of viral cis elements conferring Kaposi’s sarcoma-associated herpesvirus episome partitioning and maintenance. J. Virol. 2007, 81, 9825–9837.
- Friborg, J., Jr.; Kong, W.; Hottiger, M.O.; Nabel, G.J. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 1999, 402, 889–894.
- Krithivas, A.; Young, D.B.; Liao, G.; Greene, D.; Hayward, S.D. Human herpesvirus 8 LANA interacts with proteins of the mSin3 corepressor complex and negatively regulates Epstein-Barr virus gene expression in dually infected PEL cells. J. Virol. 2000, 74, 9637–9645.
- Lu, F.; Day, L.; Gao, S.J.; Lieberman, P.M. Acetylation of the latency-associated nuclear antigen regulates repression of Kaposi’s sarcoma-associated herpesvirus lytic transcription. J. Virol. 2006, 80, 5273–5282.
- Radkov, S.A.; Kellam, P.; Boshoff, C. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 2000, 6, 1121–1127.
- Verma, S.C.; Lan, K.; Choudhuri, T.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen modulates K1 expression through its cis-acting elements within the terminal repeats. J. Virol. 2006, 80, 3445–3458.
- Viejo-Borbolla, A.; Kati, E.; Sheldon, J.A.; Nathan, K.; Mattsson, K.; Szekely, L.; Schulz, T.F. A Domain in the C-terminal region of latency-associated nuclear antigen 1 of Kaposi’s sarcoma-associated Herpesvirus affects transcriptional activation and binding to nuclear heterochromatin. J. Virol. 2003, 77, 7093–7100.
- Allen, R.D., 3rd; Dickerson, S.; Speck, S.H. Identification of spliced gammaherpesvirus 68 LANA and v-cyclin transcripts and analysis of their expression in vivo during latent infection. J. Virol. 2006, 80, 2055–2062.
- Ebrahimi, B.; Dutia, B.M.; Roberts, K.L.; Garcia-Ramirez, J.J.; Dickinson, P.; Stewart, J.P.; Ghazal, P.; Roy, D.J.; Nash, A.A. Transcriptome profile of murine gammaherpesvirus-68 lytic infection. J. Gen. Virol. 2003, 84, 99–109.
- Forrest, J.C.; Paden, C.R.; Allen, R.D., 3rd; Collins, J.; Speck, S.H. ORF73-null murine gammaherpesvirus 68 reveals roles for mLANA and p53 in virus replication. J. Virol. 2007, 81, 11957–11971.
- Fowler, P.; Marques, S.; Simas, J.P.; Efstathiou, S. ORF73 of murine herpesvirus-68 is critical for the establishment and maintenance of latency. J. Gen. Virol. 2003, 84, 3405–3416.
- Moorman, N.J.; Willer, D.O.; Speck, S.H. The gammaherpesvirus 68 latency-associated nuclear antigen homolog is critical for the establishment of splenic latency. J. Virol. 2003, 77, 10295–10303.
- Rochford, R.; Lutzke, M.L.; Alfinito, R.S.; Clavo, A.; Cardin, R.D. Kinetics of murine gammaherpesvirus 68 gene expression following infection of murine cells in culture and in mice. J. Virol. 2001, 75, 4955–4963.
- Tsurumi, T.; Fujita, M.; Kudoh, A. Latent and lytic Epstein-Barr virus replication strategies. Rev. Med. Virol. 2005, 15, 3–15.
- Hu, J.; Yang, Y.; Turner, P.C.; Jain, V.; McIntyre, L.M.; Renne, R. LANA binds to multiple active viral and cellular promoters and associates with the H3K4methyltransferase hSET1 complex. PLoS Pathog. 2014, 10, e1004240.
- Lin, X.; Sun, R.; Zhang, F.; Gao, Y.; Bin, L.; Lan, K. The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus inhibits expression of SUMO/sentrin-specific peptidase 6 to facilitate establishment of latency. J. Virol. 2017, 91, e00806-17.
- Lu, F.; Tsai, K.; Chen, H.S.; Wikramasinghe, P.; Davuluri, R.V.; Showe, L.; Domsic, J.; Marmorstein, R.; Lieberman, P.M. Identification of host-chromosome binding sites and candidate gene targets for Kaposi’s sarcoma-associated herpesvirus LANA. J. Virol. 2012, 86, 5752–5762.
- Mercier, A.; Arias, C.; Madrid, A.S.; Holdorf, M.M.; Ganem, D. Site-specific association with host and viral chromatin by Kaposi’s sarcoma-associated herpesvirus LANA and its reversal during lytic reactivation. J. Virol. 2014, 88, 6762–6777.
- Hellert, J.; Weidner-Glunde, M.; Krausze, J.; Richter, U.; Adler, H.; Fedorov, R.; Pietrek, M.; Ruckert, J.; Ritter, C.; Schulz, T.F.; et al. A structural basis for BRD2/4-mediated host chromatin interaction and oligomer assembly of Kaposi sarcoma-associated herpesvirus and murine gammaherpesvirus LANA proteins. PLoS Pathog. 2013, 9, e1003640.
- Conrad, R.J.; Fozouni, P.; Thomas, S.; Sy, H.; Zhang, Q.; Zhou, M.M.; Ott, M. The short isoform of BRD4 promotes HIV-1 latency by engaging repressive SWI/SNF chromatin-remodeling complexes. Mol. Cell 2017, 67, 1001–1012.
- Huang, H.; Liu, S.; Jean, M.; Simpson, S.; Huang, H.; Merkley, M.; Hayashi, T.; Kong, W.; Rodriguez-Sanchez, I.; Zhang, X.; et al. A novel bromodomain inhibitor reverses HIV-1 latency through specific binding with BRD4 to Promote Tat and P-TEFb Association. Front. Microbiol. 2017, 8, 1035.
- Lu, P.; Shen, Y.; Yang, H.; Wang, Y.; Jiang, Z.; Yang, X.; Zhong, Y.; Pan, H.; Xu, J.; Lu, H.; et al. BET inhibitors RVX-208 and PFI-1 reactivate HIV-1 from latency. Sci. Rep. 2017, 7, 16646.
- Niu, Q.; Liu, Z.; Alamer, E.; Fan, X.; Chen, H.; Endsley, J.; Gelman, B.B.; Tian, B.; Kim, J.H.; Michael, N.L.; et al. Structure-guided drug design identifies a BRD4-selective small molecule that suppresses HIV. J. Clin. Investig. 2019, 129, 3361–3373.
- Akagi, T.; Ono, H.; Shimotohno, K. Characterization of T cells immortalized by Tax1 of human T-cell leukemia virus type 1. Blood 1995, 86, 4243–4249.
- Bex, F.; Gaynor, R.B. Regulation of gene expression by HTLV-I Tax protein. Methods 1998, 16, 83–94.
- Franklin, A.A.; Nyborg, J.K. Mechanisms of tax regulation of human T cell leukemia virus type I gene expression. J. Biomed. Sci. 1995, 2, 17–29.
- Grassmann, R.; Dengler, C.; Muller-Fleckenstein, I.; Fleckenstein, B.; McGuire, K.; Dokhelar, M.C.; Sodroski, J.G.; Haseltine, W.A. Transformation to continuous growth of primary human T lymphocytes by human T-cell leukemia virus type I X-region genes transduced by a Herpesvirus saimiri vector. Proc. Natl. Acad. Sci. USA 1989, 86, 3351–3355.
- Kashanchi, F.; Brady, J.N. Transcriptional and post-transcriptional gene regulation of HTLV-1. Oncogene 2005, 24, 5938–5951.
- Nerenberg, M.; Hinrichs, S.H.; Reynolds, R.K.; Khoury, G.; Jay, G. The tat gene of human T-lymphotropic virus type 1 induces mesenchymal tumors in transgenic mice. Science 1987, 237, 1324–1329.
- Tanaka, A.; Takahashi, C.; Yamaoka, S.; Nosaka, T.; Maki, M.; Hatanaka, M. Oncogenic transformation by the tax gene of human T-cell leukemia virus type I in vitro. Proc. Natl. Acad. Sci. USA 1990, 87, 1071–1075.
- Wu, X.; Qi, J.; Bradner, J.E.; Xiao, G.; Chen, L.F. Bromodomain and extraterminal (BET) protein inhibition suppresses human T cell leukemia virus 1 (HTLV-1) Tax protein-mediated tumorigenesis by inhibiting nuclear factor kappaB (NF-kappaB) signaling. J. Biol. Chem. 2013, 288, 36094–36105.
- Wang, J.; Li, G.L.; Ming, S.L.; Wang, C.F.; Shi, L.J.; Su, B.Q.; Wu, H.T.; Zeng, L.; Han, Y.Q.; Liu, Z.H.; et al. BRD4 inhibition exerts anti-viral activity through DNA damage-dependent innate immune responses. PLoS Pathog. 2020, 16, e1008429.
- Mann, M.; Roberts, D.S.; Zhu, Y.; Li, Y.; Zhou, J.; Ge, Y.; Brasier, A.R. Discovery of RSV-Induced BRD4 protein interactions using native immunoprecipitation and parallel accumulation-serial fragmentation (PASEF) mass spectrometry. Viruses 2021, 13, 454.
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123.
- Astuti, I. Ysrafil severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab. Syndr. 2020, 14, 407–412.
- Singh, S.P.; Pritam, M.; Pandey, B.; Yadav, T.P. Microstructure, pathophysiology, and potential therapeutics of COVID-19: A comprehensive review. J. Med. Virol. 2021, 93, 275–299.
- Hoesel, B.; Schmid, J.A. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86.
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A.; et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020, 26, 1636–1643.
- Wu, Z.; McGoogan, J.M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: Summary of a report of 72314 cases from the chinese center for disease control and prevention. JAMA 2020, 323, 1239–1242.
- Seltzer, S. Linking ACE2 and angiotensin II to pulmonary immunovascular dysregulation in SARS-CoV-2 infection. Int. J. Infect. Dis. 2020, 101, 42–45.
- Zumla, A.; Chan, J.F.; Azhar, E.I.; Hui, D.S.; Yuen, K.Y. Coronaviruses—Drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347.
- Woo, P.C.; Lau, S.K.; Lam, C.S.; Lau, C.C.; Tsang, A.K.; Lau, J.H.; Bai, R.; Teng, J.L.; Tsang, C.C.; Wang, M.; et al. Discovery of seven novel Mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 2012, 86, 3995–4008.
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192.
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273.
- Liu, J.; Zheng, X.; Tong, Q.; Li, W.; Wang, B.; Sutter, K.; Trilling, M.; Lu, M.; Dittmer, U.; Yang, D. Overlapping and discrete aspects of the pathology and pathogenesis of the emerging human pathogenic coronaviruses SARS-CoV, MERS-CoV, and 2019-nCoV. J. Med. Virol. 2020, 92, 491–494.
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016, 24, 490–502.
- de Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534.
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236.
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269.
- Mu, J.; Xu, J.; Zhang, L.; Shu, T.; Wu, D.; Huang, M.; Ren, Y.; Li, X.; Geng, Q.; Xu, Y.; et al. SARS-CoV-2-encoded nucleocapsid protein acts as a viral suppressor of RNA interference in cells. Sci. China Life Sci. 2020, 63, 1413–1416.
- Voss, D.; Pfefferle, S.; Drosten, C.; Stevermann, L.; Traggiai, E.; Lanzavecchia, A.; Becker, S. Studies on membrane topology, N-glycosylation and functionality of SARS-CoV membrane protein. Virol J. 2009, 6, 79.
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165878.
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.
- Xu, H.; Zhong, L.; Deng, J.; Peng, J.; Dan, H.; Zeng, X.; Li, T.; Chen, Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020, 12, 8.
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620.
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734.
- Lukassen, S.; Chua, R.L.; Trefzer, T.; Kahn, N.C.; Schneider, M.A.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A.W.; et al. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020, 39, e105114.
- Sungnak, W.; Huang, N.; Becavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687.
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574.
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904.
- Cyranoski, D. Profile of a killer: The complex biology powering the coronavirus pandemic. Nature 2020, 581, 22–26.
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263.
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Consortium, C.-G.U.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424.
- Masters, P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006, 66, 193–292.
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423.
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23.
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170.
- Mukherjee, R.; Bhattacharya, A.; Bojkova, D.; Mehdipour, A.R.; Shin, D.; Khan, K.S.; Hei-Yin Cheung, H.; Wong, K.B.; Ng, W.; Cinatl, J.; et al. Famotidine inhibits Toll-like receptor 3-mediated inflammatory signaling in SARS-CoV2 infection. J. Biol. Chem. 2021, 100925.
- Sohn, K.M.; Lee, S.G.; Kim, H.J.; Cheon, S.; Jeong, H.; Lee, J.; Kim, I.S.; Silwal, P.; Kim, Y.J.; Paik, S.; et al. COVID-19 Patients upregulate Toll-like receptor 4-mediated inflammatory signaling that mimics bacterial sepsis. J. Korean Med. Sci. 2020, 35, e343.
- Shirato, K.; Kizaki, T. SARS-CoV-2 spike protein S1 subunit induces pro-inflammatory responses via toll-like receptor 4 signaling in murine and human macrophages. Heliyon 2021, 7, e06187.
- Zhao, Y.; Kuang, M.; Li, J.; Zhu, L.; Jia, Z.; Guo, X.; Hu, Y.; Kong, J.; Yin, H.; Wang, X.; et al. SARS-CoV-2 spike protein interacts with and activates TLR4. Cell Res. 2021, 31, 825.
- Sun, Y.; Huang, J.; Song, K. BET protein inhibition mitigates acute myocardial infarction damage in rats via the TLR4/TRAF6/NF-kappaB pathway. Exp. Ther. Med. 2015, 10, 2319–2324.
- Zhao, J.; Meng, Z.; Xie, C.; Yang, C.; Liu, Z.; Wu, S.; Wang, B.; Fan, P.; Jin, X.; Wu, H. B7-H3 is regulated by BRD4 and promotes TLR4 expression in pancreatic ductal adenocarcinoma. Int. J. Biochem. Cell Biol. 2019, 108, 84–91.
- Tian, B.; Yang, J.; Zhao, Y.; Ivanciuc, T.; Sun, H.; Garofalo, R.P.; Brasier, A.R. BRD4 Couples NF-kappaB/RelA with airway inflammation and the IRF-RIG-I amplification loop in respiratory syncytial virus infection. J. Virol. 2017, 91.
- Goel, S.; Saheb Sharif-Askari, F.; Saheb Sharif Askari, N.; Madkhana, B.; Alwaa, A.M.; Mahboub, B.; Zakeri, A.M.; Ratemi, E.; Hamoudi, R.; Hamid, Q.; et al. SARS-CoV-2 Switches ‘on’ MAPK and NFkappaB signaling via the reduction of nuclear DUSP1 and DUSP5 expression. Front. Pharmacol. 2021, 12, 631879.
- Huang, B.; Yang, X.D.; Zhou, M.M.; Ozato, K.; Chen, L.F. Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol. Cell Biol. 2009, 29, 1375–1387.
- Zou, Z.; Huang, B.; Wu, X.; Zhang, H.; Qi, J.; Bradner, J.; Nair, S.; Chen, L.F. Brd4 maintains constitutively active NF-kappaB in cancer cells by binding to acetylated RelA. Oncogene 2014, 33, 2395–2404.
- Brasier, A.R.; Tian, B.; Jamaluddin, M.; Kalita, M.K.; Garofalo, R.P.; Lu, M. RelA Ser276 phosphorylation-coupled Lys310 acetylation controls transcriptional elongation of inflammatory cytokines in respiratory syncytial virus infection. J. Virol. 2011, 85, 11752–11769.
- Wang, J.; Chen, J.; Jin, H.; Lin, D.; Chen, Y.; Chen, X.; Wang, B.; Hu, S.; Wu, Y.; Wu, Y.; et al. BRD4 inhibition attenuates inflammatory response in microglia and facilitates recovery after spinal cord injury in rats. J. Cell Mol. Med. 2019, 23, 3214–3223.
- Ren, W.; Wang, C.; Wang, Q.; Zhao, D.; Zhao, K.; Sun, D.; Liu, X.; Han, C.; Hou, J.; Li, X.; et al. Bromodomain protein Brd3 promotes Ifnb1 transcription via enhancing IRF3/p300 complex formation and recruitment to Ifnb1 promoter in macrophages. Sci. Rep. 2017, 7, 39986.
- Hariharan, A.; Hakeem, A.R.; Radhakrishnan, S.; Reddy, M.S.; Rela, M. The role and therapeutic potential of NF-kappa-B pathway in severe COVID-19 Patients. Inflammopharmacology 2021, 29, 91–100.
- Xin, P.; Xu, X.; Deng, C.; Liu, S.; Wang, Y.; Zhou, X.; Ma, H.; Wei, D.; Sun, S. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int. Immunopharmacol. 2020, 80, 106210.
- Leal, A.S.; Williams, C.R.; Royce, D.B.; Pioli, P.A.; Sporn, M.B.; Liby, K.T. Bromodomain inhibitors, JQ1 and I-BET 762, as potential therapies for pancreatic cancer. Cancer Lett. 2017, 394, 76–87.
- Kleppe, M.; Koche, R.; Zou, L.; van Galen, P.; Hill, C.E.; Dong, L.; De Groote, S.; Papalexi, E.; Hanasoge Somasundara, A.V.; Cordner, K.; et al. Dual targeting of oncogenic activation and inflammatory signaling increases therapeutic efficacy in myeloproliferative neoplasms. Cancer Cell. 2018, 33, 785–787.
- Pooladanda, V.; Thatikonda, S.; Muvvala, S.P.; Devabattula, G.; Godugu, C. BRD4 targeting nanotherapy prevents lipopolysaccharide induced acute respiratory distress syndrome. Int. J. Pharm. 2021, 601, 120536.
- Ratajczak, M.Z.; Kucia, M. SARS-CoV-2 infection and overactivation of Nlrp3 inflammasome as a trigger of cytokine “storm” and risk factor for damage of hematopoietic stem cells. Leukemia 2020, 34, 1726–1729.
- Makoni, N.J.; Nichols, M.R. The intricate biophysical puzzle of caspase-1 activation. Arch. Biochem. Biophys. 2021, 699, 108753.
- Tan, Y.F.; Wang, M.; Chen, Z.Y.; Wang, L.; Liu, X.H. Inhibition of BRD4 prevents proliferation and epithelial-mesenchymal transition in renal cell carcinoma via NLRP3 inflammasome-induced pyroptosis. Cell Death Dis. 2020, 11, 239.
- Lee, C. Therapeutic modulation of virus-induced oxidative stress via the Nrf2-dependent antioxidative pathway. Oxid Med. Cell Longev. 2018, 2018, 6208067.
- Sharif-Askari, N.S.; Sharif-Askari, F.S.; Mdkhana, B.; Hussain Alsayed, H.A.; Alsafar, H.; Alrais, Z.F.; Hamid, Q.; Halwani, R. Upregulation of oxidative stress gene markers during SARS-CoV-2 viral infection. Free Radic. Biol. Med. 2021, 172, 688–698.
- Hussong, M.; Borno, S.T.; Kerick, M.; Wunderlich, A.; Franz, A.; Sultmann, H.; Timmermann, B.; Lehrach, H.; Hirsch-Kauffmann, M.; Schweiger, M.R. The bromodomain protein BRD4 regulates the KEAP1/NRF2-dependent oxidative stress response. Cell Death Dis. 2014, 5, e1195.
- Feldman, A.M.; Combes, A.; Wagner, D.; Kadakomi, T.; Kubota, T.; Li, Y.Y.; McTiernan, C. The role of tumor necrosis factor in the pathophysiology of heart failure. J. Am. Coll. Cardiol. 2000, 35, 537–544.
- Brown, J.D.; Lin, C.Y.; Duan, Q.; Griffin, G.; Federation, A.; Paranal, R.M.; Bair, S.; Newton, G.; Lichtman, A.; Kung, A.; et al. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol. Cell 2014, 56, 219–231.
- Ostuni, R.; Piccolo, V.; Barozzi, I.; Polletti, S.; Termanini, A.; Bonifacio, S.; Curina, A.; Prosperini, E.; Ghisletti, S.; Natoli, G. Latent enhancers activated by stimulation in differentiated cells. Cell 2013, 152, 157–171.
- Papait, R.; Cattaneo, P.; Kunderfranco, P.; Greco, C.; Carullo, P.; Guffanti, A.; Vigano, V.; Stirparo, G.G.; Latronico, M.V.; Hasenfuss, G.; et al. Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 20164–20169.
- Anand, P.; Brown, J.D.; Lin, C.Y.; Qi, J.; Zhang, R.; Artero, P.C.; Alaiti, M.A.; Bullard, J.; Alazem, K.; Margulies, K.B.; et al. BET bromodomains mediate transcriptional pause release in heart failure. Cell 2013, 154, 569–582.
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 receptor ACE2 Is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 2020, 181, 1016–1035.
- Alsoussi, W.B.; Turner, J.S.; Case, J.B.; Zhao, H.; Schmitz, A.J.; Zhou, J.Q.; Chen, R.E.; Lei, T.; Rizk, A.A.; McIntire, K.M.; et al. A Potently Neutralizing Antibody Protects Mice against SARS-CoV-2 Infection. J. Immunol. 2020, 205, 915–922.
- Iwata-Yoshikawa, N.; Okamura, T.; Shimizu, Y.; Hasegawa, H.; Takeda, M.; Nagata, N. TMPRSS2 contributes to virus spread and immunopathology in the airways of murine models after coronavirus infection. J. Virol. 2019, 93, e01815-18.
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkruys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado Del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. 2020, 181, 905–913.
- Tortorici, M.A.; Beltramello, M.; Lempp, F.A.; Pinto, D.; Dang, H.V.; Rosen, L.E.; McCallum, M.; Bowen, J.; Minola, A.; Jaconi, S.; et al. Ultrapotent human antibodies protect against SARS-CoV-2 challenge via multiple mechanisms. Science 2020, 370, 950–957.
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 811–818.
- Szekely, Y.; Lichter, Y.; Taieb, P.; Banai, A.; Hochstadt, A.; Merdler, I.; Gal Oz, A.; Rothschild, E.; Baruch, G.; Peri, Y.; et al. Spectrum of cardiac manifestations in COVID-19: A systematic echocardiographic study. Circulation 2020, 142, 342–353.
- Li, Y.; Zhang, Z.; Yang, L.; Lian, X.; Xie, Y.; Li, S.; Xin, S.; Cao, P.; Lu, J. The MERS-CoV receptor DPP4 as a Candidate binding target of the SARS-CoV-2 Spike. iScience 2020, 23, 101400.
- Solerte, S.B.; Di Sabatino, A.; Galli, M.; Fiorina, P. Dipeptidyl peptidase-4 (DPP4) inhibition in COVID-19. Acta Diabetol. 2020, 57, 779–783.
- Gilham, D.; Tsujikawa, L.M.; Sarsons, C.D.; Halliday, C.; Wasiak, S.; Stotz, S.C.; Jahagirdar, R.; Sweeney, M.; Johansson, J.O.; Wong, N.C.W.; et al. Apabetalone downregulates factors and pathways associated with vascular calcification. Atherosclerosis 2019, 280, 75–84.
- Jahagirdar, R.; Zhang, H.; Azhar, S.; Tobin, J.; Attwell, S.; Yu, R.; Wu, J.; McLure, K.G.; Hansen, H.C.; Wagner, G.S.; et al. A novel BET bromodomain inhibitor, RVX-208, shows reduction of atherosclerosis in hyperlipidemic ApoE deficient mice. Atherosclerosis 2014, 236, 91–100.
- Tsujikawa, L.M.; Fu, L.; Das, S.; Halliday, C.; Rakai, B.D.; Stotz, S.C.; Sarsons, C.D.; Gilham, D.; Daze, E.; Wasiak, S.; et al. Apabetalone (RVX-208) reduces vascular inflammation in vitro and in CVD patients by a BET-dependent epigenetic mechanism. Clin. Epigenetics 2019, 11, 102.
- Wasiak, S.; Gilham, D.; Daze, E.; Tsujikawa, L.M.; Halliday, C.; Stotz, S.C.; Rakai, B.D.; Fu, L.; Jahagirdar, R.; Sweeney, M.; et al. Epigenetic modulation by apabetalone counters cytokine-driven acute phase response in vitro, in mice and in patients with cardiovascular disease. Cardiovasc. Ther. 2020, 2020, 9397109.
- Lucas, J.M.; Heinlein, C.; Kim, T.; Hernandez, S.A.; Malik, M.S.; True, L.D.; Morrissey, C.; Corey, E.; Montgomery, B.; Mostaghel, E.; et al. The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discov. 2014, 4, 1310–1325.
- Belotserkovskaya, R.; Reinberg, D. Facts about FACT and transcript elongation through chromatin. Curr. Opin. Genet. Dev. 2004, 14, 139–146.
- Zhou, D.; Park, J.G.; Wu, Z.; Huang, H.; Fiches, G.N.; Biswas, A.; Li, T.W.; Ma, Q.; Martinez-Sobrido, L.; Santoso, N.; et al. FACT subunit SUPT16H associates with BRD4 and contributes to silencing of antiviral interferon signaling. bioRxiv 2021.
- Acharya, A.; Pandey, K.; Thurman, M.; Challagundala, K.B.; Vann, K.R.; Kutateladze, T.G.; Morales, G.A.; Durden, D.L.; Byrareddy, S.N. Blockade of SARS-CoV-2 infection in vitro by highly potent PI3K-alpha/mTOR/BRD4 inhibitor. bioRxiv 2021.
- Aydin, A.D.; Altinel, F.; Erdogmus, H.; Son, C.D. Allergen fragrance molecules: A potential relief for COVID-19. BMC Complement. Med. Ther. 2021, 21, 41.
- Liskova, A.; Samec, M.; Koklesova, L.; Samuel, S.M.; Zhai, K.; Al-Ishaq, R.K.; Abotaleb, M.; Nosal, V.; Kajo, K.; Ashrafizadeh, M.; et al. Flavonoids against the SARS-CoV-2 induced inflammatory storm. Biomed. Pharmacother. 2021, 138, 111430.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
2 times
(View History)
Update Date:
05 Aug 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No