Phosphodiesterases (PDEs) hydrolyze cyclic nucleotides to modulate multiple signaling events in cells. PDEs are recognized to actively associate with cyclic nucleotide receptors (protein kinases, PKs) in larger macromolecular assemblies referred to as signalosomes. Complexation of PDEs with PKs generates an expanded active site that enhances PDE activity. This facilitates signalosome-associated PDEs to preferentially catalyze active hydrolysis of cyclic nucleotides bound to PKs and aid in signal termination. PDEs are important drug targets, and current strategies for inhibitor discovery are based entirely on targeting conserved PDE catalytic domains. This often results in inhibitors with cross-reactivity amongst closely related PDEs and attendant unwanted side effects.
1. Introduction
Second-messenger cyclic nucleotides (cNMPs) are important regulators of numerous cellular pathways. Phosphodiesterases (PDEs) catalyze the hydrolysis of cyclic nucleotides regulating the overall levels of cyclic nucleotides and thereby impact the magnitude and duration of the cellular response. This makes them important targets for drug discovery
[1][2][3]. The PDE superfamily comprises 11 different families in mammals, each with numerous subtypes and isoforms
[4]. Isoforms of various PDEs have been effectively targeted to treat cardiac arrhythmia, inflammation, erectile dysfunction, and steroidogenesis
[1]. Based on their substrate specificity, PDEs are categorized broadly into cyclic 3′, 5′ adenosine monophosphate (cAMP)- and cyclic 3′, 5′ guanosine monophosphate (cGMP)- specific and dual-specificity PDEs. About 100 PDEs are thus distributed in various tissues and across different stages of development. Each PDE isoform includes a conserved C-terminal catalytic domain associated with one or more variable N-terminal regulatory domains. Association of PDEs with specific receptors and cyclases to form signaling islands referred to as ‘signalosomes’ has been increasingly recognized to be the primary mode of cyclic nucleotide regulation in cells
[5][6][7]. These signaling islands are mediated by specific scaffold proteins that localize multiple elements of the cNMP signaling pathway
[8][9][10][11][12][13][14].
In signalosomes, PDEs are anchored in close proximity to cyclic nucleotide receptors and function as multivalent macromolecular assemblies, rather than as free diffusive PDEs
[6][15]. An imperative effect of such colocalization of PDEs and cNMP receptors is that the PDEs are poised to hydrolyze cNMP bound to the receptors, i.e. protein kinases (PKs)
[16]. This ‘direct’ hydrolysis of bound cNMPs offers precision in regulating various signaling pathways and imparts specificity. Inhibition of PDEs leads to an overall increase in cNMP levels, which is essential in disease control. Due to the ubiquitous presence of PDEs in the body, low selectivity of particular inhibitors to their respective PDEs gets translated into unintended adverse effects in cells. For example, cyanopsia (i.e., blue tinge in vision) is associated with the use of sildenafil, a PDE5-specific inhibitor. This visual symptom occurs due to its cross-reactivity of sildenafil with PDE6, which is present only in rod and cone photoreceptors
[17][18]. Hence, this necessitates more isoform-specific inhibitors to overcome the cross-reactivity problem faced by current PDE inhibitors. Targeting the receptor-bound PDE, rather than PDEs alone, overcomes some of the limitations of nonspecific overlapping effects that inhibitors targeting conserved PDE sites might present.
Our goal was to determine the degree of PDE inhibition by different small molecules using PDE–PK complexes as the targets. To screen inhibition of cAMP- or cGMP-specific PDEs, we selectively chose (i) cAMP-specific- PDE8-protein kinase A complex (mammalian), (ii) cGMP-specific- PDE5-protein kinase G (PKG) complex (mammalian), and (iii) a dual cAMP/cGMP selective RegA–PKA system (from
Dictyostelium discoideum). cAMP-dependent protein kinase A (PKA) and cGMP-dependent protein kinase G (PKG) represent two of the most important cyclic nucleotide effectors
[15][19]. Regulatory subunit of PKA (‘PKAR’) and PKG have two non-redundant high-affinity cyclic nucleotide binding (CNB) sites
[20][21][22]. PDEs couple to these bound cyclic nucleotides to form PDE–PK complexes with ‘composite active sites’
[6][23]. Given that most intracellular PDEs are localized within signalosomes
[8][11][13], the PDE–PK complexes represent relevant high-specificity targets for inhibitor discovery.
2. Designing Competitive cNMP-Dependent Displacement Assay for PDE Inhibitors
We first set out to assess a phosphodiesterase–protein-kinase complex as a tool to monitor the hydrolysis of cyclic nucleotides and its displacement by small molecules. To monitor real-time association or dissociation of cyclic nucleotides, we used fluorescent analogs of cAMP (2fluo-cAMP, ‘2fc’) and cGMP (2fluo-cGMP, ‘2fg’). Using fluorescence polarization (FP), we first measured the kinetic complexation of catalytic domains of PDEs with their specific protein kinases and tested their stability using cAMP or cGMP as substrates.
2.1. Designing a cAMP-Specific Assay Using PDE8–PKAR Complex
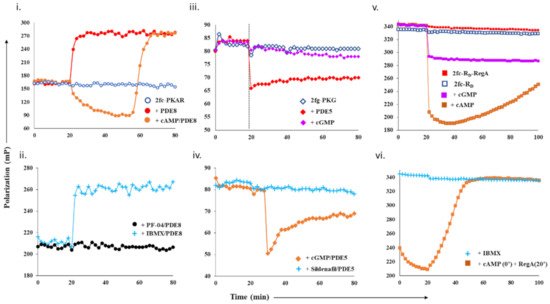
For cAMP-specific PDEs, we used the mammalian PDE8–PKAR system. FP of 2fluo-cAMP-bound PKAR (‘2fc-PKAR’) was constant throughout (i, blue), indicating their stable binding. Next, PDE8c in the absence or presence of excess cAMP was added at time t = 20 min. Addition of PDE8c led to an immediate increase in FP values (red plot), suggesting the formation of a complex between PDE8c and 2fc-PKAR, which remained stable over time. Addition of cAMP-saturated PDE8c led to a gradual decrease in FP (orange plot), which indicates competitive displacement of 2fluo-cAMP from PKAR by cAMP. The decrease in FP was followed by a rapid increase in FP, corresponding to those measured for the 2fc-PKAR–PDE8c complex rather than 2fc-PKAR. Such an FP trend suggests that the PDE–PKAR composite active site preferentially hydrolyzed unlabeled cAMP first, followed by reassociation of the 2fc-PKAR–PDE8c complex.
Figure 1. Design and test of competitive displacement assay using PDE–PK complexes. (i,ii) cAMP-specific probe PDE8–PKAR: (i) active PDE8c (red circles) and cAMP-saturated PDE8c (orange circles) were added to 2fc-PKAR at 20 min interval, and the FP values were measured; (ii) inhibition and complexation of cAMP-specific PDE8–PKAR were tested by the addition of PDE8c (1 µM) incubated with 10 µM PF-04957325 (black circles) and 500 µM IBMX (cyan plus) to 2fc-PKAR at t = 20 min. (iii,iv) cGMP-specific probe PDE5–PKG: (iii) at 20 min time interval, PDE5 (red diamonds) and cGMP (lilac diamonds) were added to 2fg-PKG (blue circles), and the signal was measured for a total time course of 80 min; (iv) PDE5 was incubated with substrate 100 µM cGMP (orange diamonds) and 1 µM sildenafil (cyan plus) and added to 2fg-PKG at time t = 30 min to test stability and inhibition of cGMP-specific PDE5–PKG probe. (v,vi) Dual-specificity probe RegA-RD: (v) Plot showing stability of preformed composite active sites of 2fc-RD-RegA (red squares) as compared to free 2fc-RD (blue squares). Excess cAMP (orange squares) and cGMP (lilac squares) were added to 2fc-RD-RegA complex at 20 min interval and FP measured for further 80 min. (vi) At t = 20 min, IBMX (cyan plus) was added to 2fc-RD. Separately, cAMP was added to 2fc-RD at time = 0 min, followed by RegA at time = 20 min, to this reaction mixture (orange squares), and FP was measured for a time course of 100 min.
To use the PDE8–PKAR complex as a tool to screen novel inhibitors, we first tested the complex with two known inhibitors—IBMX and PF-04957325. IBMX is a broad-specific PDE inhibitor IBMX, which binds PDE8 but does not inhibit it, while PF-04957325 is a PDE8-specific inhibitor (IC
50~1–30 nM)
[24][25]. PDE8c incubated with IBMX and PF-04957325 was added to 2fc-PKAR to test for inhibition and complexation (ii). Consistent with our expectations, we observed that the addition of IBMX-saturated PDE8c (cyan plus plots) resulted in increased FP values similar to those measured for active PDE8c (red plot, i). This indicated that IBMX did not inhibit PDE8c, which led to 2fc-PKAR–PDE8c complex formation. Our FP results show no reduction in FP for IBMX-treated PDE8, suggesting that intracellular PDE8 preferred to be associated with cAMP receptors as a composite catalytic site, and therefore preventing PDE8 inhibition by IBMX. This is consistent with literature where IBMX is shown to inhibit other PDEs, but not PDE8, although high-resolution crystal structure showed IBMX-bound PDE8
[26][27]. Addition of PDE8c incubated with PF-04957325 (black circles plot) resulted in no significant change, where FP values remained equivalent to 2fc-PKAR (blue open circles, i). Lack of any increase in FP indicated that PF-04957325-inhibited PDE8c was unable to bind to 2fc-PKAR. These results indicate that PDE8–PKAR-composite-site-based fluorescence polarization assay offered a reliable platform to identify inhibitors and distinguish them from small molecules that occupy the catalytic sites.
2.2. Designing Assay for cGMP-Specificity Using PDE5–PKG Complex
We next determined the complexation of cGMP-specific PDE5 with PKG and monitored the binding of cGMP and PDE5 to 2fg-PKG (iii, blue diamonds). PDE5 addition led to an instant drop in FP (red plot). These results suggest that the PDE5–PKG complex mediated hydrolysis of substrate 2fluo-cGMP, followed by PDE5 dissociation leading to decreased FP. Meanwhile, addition of 1000-fold excess cGMP (lilac plot) resulted in no significant change in FP, indicating that 2fluo-cGMP remained stably bound to PKG. We then set out to determine the effect of excess cGMP on the PDE5–PKG complex. Addition of cGMP-saturated PDE5 (orange diamonds, iv) led to a decrease in FP values greater than that observed for active PDE5 alone (red diamonds, iii), followed by gradual increased FP equivalent to that of 2fg-PKG–PDE5 mixture. This drop in polarization to lower values indicated cGMP–PDE5 displaced 2fluo-cGMP from PKG, suggestive of processive hydrolysis of cGMP by PDE5–PKG composite site. The hydrolysis product 5′- GMP gets displaced by high-affinity binding 2fluo-cGMP, resulting in increased FP at the end of the reaction.
Subsequently, we tested the substrate displacement from 2fg-PKG by PDE5 in the presence of sildenafil, a PDE5-specific inhibitor
[17]. Addition of sildenafil-treated PDE5 did not result in any significant change in FP values, indicative of inhibition of PDE5. These results, therefore, highlight the specificity of using the PDE5–PKG complex as a tool to differentiate between a kinetically active composite site from an inhibited complex.
2.3. Designing a Dual cAMP/cGMP Assay Using a Broader Specificity RegA–RD Complex
As PDE catalytic domains are conserved with high structural similarity, we selected dual-specific cAMP/cGMP selective PDEs. Type 2 phosphodiesterase from
D. discoideum, RegA, has high similarity to PDE8 catalytic site and therefore was a good choice
[28][29]. In addition, RegA is not specific to cAMP and shows cAMP/cGMP selectivity of ~200, indicating it can bind to both cAMP and cGMP. Various studies have shown that RegA regulates the functions of its cognate PKA regulatory subunit (R
D), which also has two cyclic nucleotide binding sites—CNB:A (high affinity) and CNB:B (low affinity)
[19]. Here, our strategy was to differentiate small molecules that displace cAMP and/or cGMP.
We first monitored polarization of the RegA–RD complex (red square) and compared it with FP values of 2fc-RD alone (blue square plot). FP values for the 2fc-RD–RegA complex were higher than 2fc-RD, indicating stable complex formation (v). We then added excess cAMP and cGMP to the 2fc-RD–RegA complex to track the association–dissociation changes accompanying substrate hydrolysis. Addition of cAMP (orange squares) resulted in an immediate decrease in FP, indicative of competitive displacement of 2fluo-cAMP by cAMP. Importantly, we observed a gradual and steady increase in FP over the time measured, suggesting slow reassociation of 2fluo-cAMP to RD or the RegA–RD complex. While cAMP addition lead to complete displacement, we noticed that addition of cGMP (lilac plot) resulted in only a partial decrease in FP that remained stable over time. We believe that cGMP displaced 2fluo-cAMP only from one of the two composite sites, probably the low-affinity site of RD and RegA. These results served as controls for displacement from one (like cGMP) or both (like cAMP) composite sites of the dual-specificity PDE–PK complex. Next, we monitored the effects of broad-specificity PDE inhibitor IBMX on the 2fc-RD–RegA complex and observed no change in FP (cyan plus plot, vi). To simulate intracellular complex formation, cAMP was added to 2fc-RD at 0 min (orange square plot, vi), and then RegA at 20 min. Large-scale decrease in FP was observed, followed by increased FP in the presence of RegA. These trends suggested dissociation and reassociation of 2fluo-cAMP from the RegA–RD complex. Here, we developed a competitive displacement assay using the PDE–PK complex as a screening tool to distinguish between cAMP and cGMP specific inhibitors.
3. Screening Novel Inhibitors in Plant Extracts Using Competitive Displacement Assay
One of the major sources of drugs, especially PDE inhibitors, is natural sources such as plants
[30][31][32][33]. Natural products are intrinsically useful in drug discovery due to their high level of structural as well as chemical diversity
[34]. They also possess a unique advantage of having high biochemical specificity and binding affinities to their receptors
[34]. In this study, the PDE inhibitory potentials of natural products extracted from two medicinal plant species—
Swietenia macrophylla and
Vitex trifolia—were investigated using the proposed assay. Both these plants have been shown to possess phytochemicals that inhibit PDEs and other enzymes
[35]. Profiling of the phytoconstituents in these plant extracts by GC-MS lead to the identification of a wide variety of compounds of differing polarities such as fatty acids, flavonoids, phenolics, methylxanthines, glycosides, and diterpenes. We used the PDE–PK-complex-based fluorescence polarization assay to screen and identify inhibitors. As the fluorescent ligands are the reporters, any competition to their binding to the PDE–PK complex could be monitored easily and rapidly. Further, known PDE inhibitors were used to test the reliability of this assay.
3.1. Screening cAMP-Specific Inhibitors Using PDE8–PKAR Complex
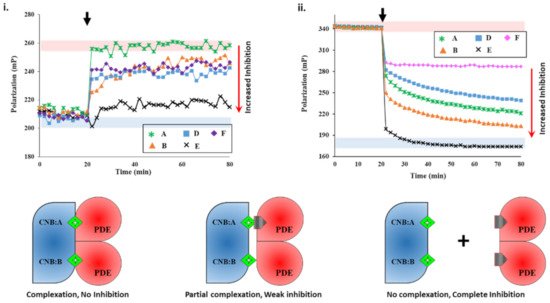
We next set out to screen plant extracts for PDE inhibition and their effects on the PDE–PK complex stability. FP results of IBMX- and PF-04957325 showed the specificity of the assay. To determine if an unknown small molecule was an inhibitor, we used active PDE8 (red area, i) and PF-04957325-inhibited PDE8 (blue area, i) serving as negative and positive controls for inhibition. First, PDE8 was incubated with 5 µL each of extracts A-F for 15 min and then added to 2fc-PKAR individually at a 20 min interval. The polarization values observed for extract-A-treated PDE8 (i) were similar to those observed for the 2fc-PKAR–PDE8 complex, indicating that the components of extract A were unable to bind or inhibit PDE8 or the PDE8–PKAR composite site. For PDE8 treated with extracts B, D, and E, the FP results observed were in-between active or inhibited composite sites, indicating that these extracts may possess phytoconstituents that block the active site partially (extracts B, D, and E), resulting in a mixture of active and inhibited PDE8–PKAR complexes. Addition of extract-E-treated PDE8 (black cross plot, i) resulted in FP values parallel to PF-04957325 (black circles plot, i). This suggests that extract E has compounds that bind and block PDE8–PKAR composite site formation and thereby inhibiting its activity. Importantly, these results depict the sensitivity of the assay to screen inhibitors of varying strengths in a single-step procedure. Therefore, the 2fc-PKAR–PDE8 complex can be used as a probe to screen small molecules that may either activate or inhibit phosphodiesterases.
Figure 2. Plant extracts inhibit PDE8 and RegA with different potencies. (i) Changes in fluorescence polarization (y-axis) over time (x-axis) are represented for 2fluo-cAMP-saturated PKAR upon addition of PDE8c pre-incubated with extracts A (green asterisk), B (orange triangles), D (blue squares), E (black cross), and F (lilac diamonds) at 20 min (black arrow). (ii) FP plot showing effect of inhibitors on RegA-RD complexation. After 20 min (black arrow), 5 µL of extracts A, B, D, E, and F were added to preformed 2fc-RD-RegA complex. Red area indicates zone of no PDE inhibition as per negative control without any extract added, while blue area indicates high PDE inhibition zone using PDE inhibitors as positive control. (C) 2fluo-cAMP (green diamond) is bound to cyclic nucleotide binding (CNB) domains CNB:A and CNB:B of PKA (blue). Cartoons showing inhibitor (gray pentagon) mediated complete (left), partial (center), or no (right) complex formation between PDE (red) and PK (blue) due to no, partial, or complete PDE inhibition, respectively.
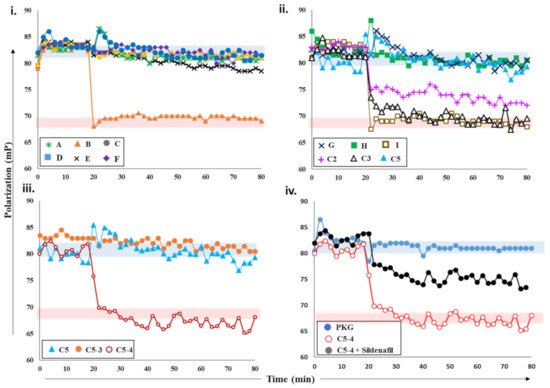
Figure 3. Effects of plant extracts and fractions on PDE5 activity. (i) Fluorescence polarization versus time plot depicting the changes in FP values of 2fg-PKG upon addition of PDE5 (0.2 µM) incubated with crude plant extracts A-F at 20 min time interval (black arrow). Red zone indicates values similar to active PDE5 (no inhibition), while blue zone indicates FP values similar to inhibited PDE5. Increased FP indicates greater FP inhibition (red arrow). Inhibition of PDE5 incubated with fractions C2, C3, C5 from crude extract C; sub-fractions C5-3 and C5-4 (panel (iii)); and pure compounds G, H, and I (panel (ii)). Red area indicates zone of no PDE inhibition based on negative-control active PDE5, while blue area indicates high PDE inhibition zone based on positive-control sildenafil. (iv) PDE5 was incubated with sub-fraction C5-4 spiked with 100 nM sildenafil (black circles) and added to 2fg-PKG (blue circles) at time t = 20 min. Sub-fraction C5-4, which does not inhibit PDE5, is shown for reference.
3.2. Screening Plant Extracts for Dual cAMP/cGMP PDE Inhibition of the RegA–RD Complex
We next screened the inhibition potency of the plant extracts against cAMP and/or cGMP binding PDEs. At 20 min intervals, the plant extracts were added to RegA–RD complex and their FP was recorded for additional 80 min. Amongst the various plant extracts tested, certain crude extracts showed no change in FP values for the RegA–RD pair (data not shown), while extracts A-F showed significant decreases in polarization values (ii). Closer inspection of FP of various extracts highlighted differences in their degree of PDE inhibition. Addition of extract E (black cross, ii) to 2fc-RD–RegA resulted in a sharp drop in FP, which indicated the rapid displacement of 2fluo-cAMP. Notably, the dissociation kinetics were faster than that observed for cAMP (v), suggesting that extract E is highly potent with phytoconstituents that bind to PDE–PK composite site with affinity greater than cAMP.
Addition of extract A showed a gradual decrease in FP values, although not as potent as extract E, indicating extract A inhibited RegA–RD. Interestingly, extract A showed no inhibition of PDE8, which suggests its variability in inhibition of cAMP–PDEs. Addition of extract F (ii) led to displacement only from one site, as the FP values were similar to those observed for cGMP (v). The potency or the ability of these compounds as potential inhibitors was observed to be E > B > A > D > F in decreasing order, where extract E showed complete and rapid displacement of 2fluo-cAMP, while extract F had the smallest effect. These results highlight the importance of targeting the PDE–PKAR complex as the ‘new active site,’ as it offers insights into the specificity and selectivity of a particular compound.
3.3. Screening cGMP-Specific Inhibitors by Targeting PKG–PDE5 Complexes
Next, we screened various plant extracts with a cGMP-specific PDE5–PKG complex. PDE5 treated with sildenafil was used as a positive control to measure PDE5 inhibition (blue area, ), while active PDE5 (red area) as a control for no inhibition. PDE5 was incubated with various crude plant extracts A-F for 15 min and then added to 2fc-PKG (i). Addition of PDE5 treated with extract B (orange triangles) resulted in decreased FP values, similar to active PDE5, while PDE5 treated with other extracts showed no significant decrease in the polarization values and was comparable to sildenafil. These observations suggested that extract B was unable to inhibit PDE5, but other extracts likely consisted of compounds that inhibit PDE5 and the PDE5–PKG composite site. The inhibition potency of these extracts was C = F > A > D > E in decreasing order.
Further characterization of the inhibition of these extracts was carried out using PDE-GloTM phosphodiesterase assay. The results of FP assay and PDE-GloTM assay were similar (described in a later section). The two complementary methods allowed us to select active extracts and further downstream processing. Crude extract C was then subjected to fractionation and separation into less complex mixtures. This yielded five fractions C1-5, which were then tested for PDE inhibition. PDE5 incubated with extracts C2, C3, and C5 was added to 2fg-PKG and their polarization values recorded (ii). Addition of C2-incubated PDE5 led to a decrease in FP values intermediate to those of active and inhibited PDE5, indicating partial/weak inhibition. When C3-incubated PDE5 was added, a greater decrease in FP was observed with values similar to active PDE5, indicative of no PDE inhibition. Fraction C5 (blue triangle) showed the greatest PDE5 inhibition as reflected by the lack of any changes in FP when mixed with 2fg-PKG.
Subsequently, fraction C5 was subjected to further fractionation to isolate the active compound(s). Fractions C5-3 and C5-4 obtained were then targeted. PDE5 treated with extract C5-4 addition resulted in a decrease in FP (maroon, open circles) with values similar to those of active PDE5. Fraction-C5-3-treated PDE5 addition led to a significant change in FP, with values similar to those observed for 2fg-PKG. These results indicate that sub-fraction C5-4 did not inhibit PDE5, while C5-3 inhibited PDE5 similar to sildenafil. Furthermore, preliminary mass spectrometry analysis (
Supplementary Figure S2iiC) of C5-3 fraction led to identification of few components which could possibly be PDE5 inhibitors. We then used commercially available standards G, H, and I of the identified small molecules. We applied our competitive displacement assay using the PDE5–PKG composite site to identify which of these compounds showed inhibition. PDE5 was incubated with three compounds G, H, and I and added to 2fg-PKG. Decreased polarization was observed for compound-I-incubated PDE5, suggesting that this did not inhibit PDE5. FP values for PDE5 treated with compounds G and H did not change over the time course of the experiment, indicating that these two were potential PDE5 inhibitors.
Current methods employed for the screening and detection of PDE5 inhibitors and their analogs as adulterants in herbal products include HPLC-UV, MS-based methods (e.g., LC-MS, GC-MS), vibrational spectroscopic methods (e.g., IR, Raman spectroscopy), and NMR spectroscopy. However, such targeted, structure-based techniques are limited by the need for prior knowledge of the chemical structure and thus have limited use. In contrast, the developed FP assay is an untargeted activity-based bioassay where detection of any adulterants is based on their pharmacological mechanism (i.e., PDE5 inhibition). Thus, its utility as a qualitative screening assay for the detection of PDE5 inhibitor analogs in natural products was investigated. As can be seen from iv, addition of PDE5 pretreated with C5-4 alone resulted in a drop in FP values similar to that of active PDE5. This indicates that C5-4 is unable to inhibit PDE5 and is in line with previous results. On the other hand, addition of C5-4 spiked with the sildenafil led to a slight drop in FP values that were between that of active and inactive PDE5. This suggests that the extract spiked with sildenafil partially inhibits PDE5. Therefore, the results indicate that the detection capability of the developed FP assay toward the sildenafil was retained even in a complex matrix such as a semi-purified fraction from a plant extract and may potentially be applied for the screening of herbal products adulterated with PDE5 inhibitors in the future.
PDE–PK composite-site-based assay is capable of rapidly distinguishing an inhibitor from a non-binding molecule. We believe that this approach has enormous implications in identifying novel compounds that bind to both kinase and phosphodiesterase and can be used as a high-throughput screening procedure.
 +1 credit
+1 credit