 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Teresa Tsang | + 3437 word(s) | 3437 | 2021-06-21 08:54:15 | | | |
| 2 | Peter Tang | Meta information modification | 3437 | 2021-06-22 04:25:08 | | |
Video Upload Options
Fabry disease (FD) is an X-linked lysosomal storage disorder caused by mutations in the galactosidase A (GLA) gene that result in deficient galactosidase A enzyme and subsequent accumulation of glycosphingolipids throughout the body. The result is a multi-system disorder characterized by cutaneous, corneal, cardiac, renal, and neurological manifestations. Increased left ventricular wall thickness represents the predominant cardiac manifestation of FD. As the disease progresses, patients may develop arrhythmias, advanced conduction abnormalities, and heart failure.
1. Introduction
2. Clinical Presentation of Fabry Disease
3. Fabry Cardiomyopathy
|
Structural abnormalities detected by cardiac imaging |
|
|
Electrophysiologic abnormalities detected by ECG or prolonged rhythm monitoring |
|
|
Patient History |
ECG |
Echocardiography |
CMR |
|
|---|---|---|---|---|
|
Fabry Cardiomyopathy |
|
|
|
|
|
Hypertension |
|
|
|
|
|
Athlete’s Heart |
|
|
|
|
|
Aortic Stenosis |
|
|
|
|
|
Hypertrophic Cardiomyopathy |
|
|
|
|
|
Cardiac Amyloidosis |
|
|
|
|
4. Screening and Diagnosis
5. Diagnosis of Fabry Cardiomyopathy
5.1. Echocardiography
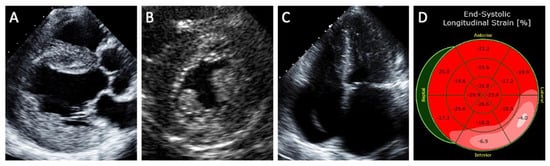
5.2. Magnetic Resonance Imaging
5.3. Laboratory Tests
5.4. Cardiopulmonary Exercise Test
6. Treatments in Fabry Disease
|
Disease-Modifying Therapy |
Advantages |
Disadvantages |
|---|---|---|
|
First-generation ERT
|
|
|
|
Oral chaperone therapy
|
|
|
|
Second-generation ERT
|
|
|
|
Substrate reduction therapy
|
|
|
|
Gene therapy |
|
|
|
Structural abnormalities that can be present on cardiac imaging |
|
|
Electrophysiologic abnormalities detected by ECG or rhythm monitoring |
|
|
Other cardiovascular considerations in patients with Fabry disease |
|
Abbreviations: ACE, angiotensin converting enzyme; ARB, angiotensin II receptor blocker; AV, atrioventricular; ERT, enzyme replacement therapy; ICD, implantable cardioverter-defibrillator; LV, left ventricular; LVEF, left ventricular ejection fraction; LVOT, left ventricular outflow tract.
References
- Germain, D.P. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30.
- Hwu, W.L.; Chien, Y.H.; Lee, N.C.; Chiang, S.C.; Dobrovolny, R.; Huang, A.C.; Yeh, H.Y.; Chao, M.C.; Lin, S.J.; Kitagawa, T.; et al. Newborn screening for fabry disease in taiwan reveals a high incidence of the later-onset GLA mutation c.936 + 919G > A (IVS4 + 919G > A). Hum. Mutat. 2009, 30, 1397–1405.
- Spada, M.; Pagliardini, S.; Yasuda, M.; Tukel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R.J. High incidence of later-onset Fabry disease revealed by newborn screening. Am. J. Hum. Genet. 2006, 79, 31–40.
- Yeung, D.F.; Sirrs, S.; Tsang, M.Y.C.; Gin, K.; Luong, C.; Jue, J.; Nair, P.; Lee, P.K.; Tsang, T.S.M. Echocardiographic Assessment of Patients with Fabry Disease. J. Am. Soc. Echocardiogr. 2018, 31, 639–649.e2.
- Pieroni, M.; Moon, J.C.; Arbustini, E.; Barriales-Villa, R.; Camporeale, A.; Vujkovac, A.C.; Elliott, P.M.; Hagege, A.; Kuusisto, J.; Linhart, A.; et al. Cardiac Involvement in Fabry Disease: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2021, 77, 922–936.
- Seydelmann, N.; Wanner, C.; Störk, S.; Ertl, G.; Weidemann, F. Fabry disease and the heart. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 195–204.
- Chamoles, N.A.; Blanco, M.; Gaggioli, D. Fabry disease: Enzymatic diagnosis in dried blood spots on filter paper. Clin. Chim. Acta 2001, 308, 195–196.
- Mehta, A.; Clarke, J.T.R.; Giugliani, R.; Elliott, P.; Linhart, A.; Beck, M.; Sunder-Plassmann, G. Natural course of Fabry disease: Changing pattern of causes of death in FOS-Fabry Outcome Survey. J. Med. Genet. 2009, 46, 548–552.
- Linhart, A.; Germain, D.P.; Olivotto, I.; Akhtar, M.M.; Anastasakis, A.; Hughes, D.; Namdar, M.; Pieroni, M.; Hagège, A.; Cecchi, F.; et al. An expert consensus document on the management of cardiovascular manifestations of Fabry disease. Eur. J. Heart Fail. 2020, 22, 1076–1096.
- Nakao, S.; Takenaka, T.; Maeda, M.; Kodama, C.; Tanaka, A.; Tahara, M.; Yoshida, A.; Kuriyama, M.; Hayashibe, H.; Sakuraba, H.; et al. An Atypical Variant of Fabry’s Disease in Men with Left Ventricular Hypertrophy. N. Engl. J. Med. 1995, 333, 288–293.
- Hsu, T.R.; Hung, S.C.; Chang, F.P.; Yu, W.C.; Sung, S.H.; Hsu, C.L.; Dzhagalov, I.; Yang, C.F.; Chu, T.H.; Lee, H.J.; et al. Later Onset Fabry Disease, Cardiac Damage Progress in Silence: Experience With a Highly Prevalent Mutation. J. Am. Coll. Cardiol. 2016, 68, 2554–2563.
- Shah, J.S.; Hughes, D.A.; Sachdev, B.; Tome, M.; Ward, D.; Lee, P.; Mehta, A.B.; Elliott, P.M. Prevalence and clinical significance of cardiac arrhythmia in Anderson-Dabry disease. Am. J. Cardiol. 2005, 96, 842–846.
- Weidemann, F.; Sanchez-Niño, M.D.; Politei, J.; Oliveira, J.P.; Wanner, C.; Warnock, D.G.; Ortiz, A. Fibrosis: A key feature of Fabry disease with potential therapeutic implications. Orphanet J. Rare Dis. 2013, 8, 1–12.
- Omahony, C.; Coats, C.; Cardona, M.; Garcia, A.; Calcagnino, M.; Murphy, E.; Robin, L.; Atul, M.; Derralynn, H.; Perry, M.E. Incidence and predictors of anti-bradycardia pacing in patients with Anderson-Fabry disease. Europace 2011, 13, 1781–1788.
- Namdar, M. Electrocardiographic Changes and Arrhythmia in Fabry Disease. Front. Cardiovasc. Med. 2016, 3, 7.
- Niemann, M.; Breunig, F.; Beer, M.; Herrmann, S.; Strotmann, J.; Hu, K.; Emmert, A.; Voelker, W.; Ertl, G.; Wanner, C.; et al. The right ventricle in Fabry disease: Natural history and impact of enzyme replacement therapy. Heart 2010, 96, 1915–1919.
- Kampmann, C.; Linhart, A.; Baehner, F.; Palecek, T.; Wiethoff, C.M.; Miebach, E.; Whybra, C.; Gal, A.; Bultas, J.; Beck, M. Onset and progression of the Anderson-Fabry disease related cardiomyopathy. Int. J. Cardiol. 2008, 130, 367–373.
- Mehta, J.; Moller, J.H.; Desnick, R.J.; Ph, D. Electrocardiographic and vectorcardiographic abnormalities in Fabry’s disease. Am. Heart J. 1977, 93, 699–705.
- Linhart, A.; Paleček, T.; Bultas, J.; Ferguson, J.J.; Hrudová, J.; Karetová, D.; Zeman, J.; Ledvinová, J.; Poupětová, H.; Elleder, M.; et al. New insights in cardiac structural changes in patients with Fabry’s disease. Am. Heart J. 2000, 139, 1101–1108.
- Barbey, F.; Qanadli, S.D.; Juli, C.; Brakch, N.; Palaek, T.; Rizzo, E.; Jeanrenaud, X.; Eckhardt, B.; Linhart, A. Aortic remodelling in Fabry disease. Eur. Heart J. 2010, 31, 347–353.
- Desnick, R.J.; Blieden, L.C.; Sharp, H.L.; Hofschire, P.J.; Moller, J.H. Cardiac valvular anomalies in Fabry disease. Clinical, morphologic, and biochemical studies. Circulation 1976, 54, 818–825.
- Sachdev, B.; Takenaka, T.; Teraguchi, H.; Tei, C.; Lee, P.; McKenna, W.J.; Elliott, P.M. Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation 2002, 105, 1407–1411.
- Maron, M.S.; Xin, W.; Sims, K.B.; Butler, R.; Haas, T.S.; Rowin, E.J.; Desnick, R.J.; Maron, B.J. Identification of Fabry Disease in a Tertiary Referral Cohort of Patients with Hypertrophic Cardiomyopathy. Am. J. Med. 2018, 131, 200.e1–200.e8.
- Doheny, D.; Srinivasan, R.; Pagant, S.; Chen, B.; Yasuda, M.; Desnick, R.J. Fabry Disease: Prevalence of affected males and heterozygotes with pathogenic GLA mutations identified by screening renal, cardiac and stroke clinics, 1995–2017. J. Med. Genet. 2018, 55, 261–268.
- Lin, H.Y.; Chong, K.W.; Hsu, J.H.; Yu, H.C.; Shih, C.C.; Huang, C.H.; Lin, S.J.; Chen, C.H.; Chiang, C.C.; Ho, H.J.; et al. High incidence of the cardiac variant of fabry disease revealed by newborn screening in the Taiwan Chinese population. Circ. Cardiovasc. Genet. 2009, 2, 450–456.
- Baig, S.; Edward, N.C.; Kotecha, D.; Liu, B.; Nordin, S.; Kozor, R.; Moon, J.C.; Geberhiwot, T.; Steeds, R.P. Ventricular arrhythmia and sudden cardiac death in Fabry disease: A systematic review of risk factors in clinical practice. Europace 2018, 20, f153–f161.
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with Fabry disease. Clin. Genet. 2016, 89, 44–54.
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of lysosomal storage disorders. J. Am. Med. Assoc. 1999, 281, 249–254.
- Mundigler, G.; Gaggl, M.; Heinze, G.; Graf, S.; Zehetgruber, M.; Lajic, N.; Voigtlander, T.; Mannhalter, C.; Sunder-Plassmann, R.; Paschke, E.; et al. The endocardial binary appearance (‘binary sign’) is an unreliable marker for echocardiographic detection of Fabry disease in patients with left ventricular hypertrophy. Eur. J. Echocardiogr. 2011, 12, 744–749.
- Gruner, C.; Verocai, F.; Carasso, S.; Vannan, M.A.; Jamorski, M.; Clarke, J.T.R.; Care, M.; Iwanochko, R.M.; Rakowski, H. Systolic myocardial mechanics in patients with Anderson-Fabry disease with and without left ventricular hypertrophy and in comparison to nonobstructive hypertrophic cardiomyopathy. Echocardiography 2012, 29, 810–817.
- Labombarda, F.; Saloux, E.; Milesi, G.; Bienvenu, B. Loss of base-to-apex circumferential strain gradient: A specific pattern of Fabry cardiomyopathy? Echocardiography 2017, 34, 504–510.
- Hindieh, W.; Weissler-Snir, A.; Hammer, H.; Adler, A.; Rakowski, H.; Chan, R.H. Discrepant Measurements of Maximal Left Ventricular Wall Thickness Between Cardiac Magnetic Resonance Imaging and Echocardiography in Patients With Hypertrophic Cardiomyopathy. Circ. Cardiovasc. Imaging 2017, 10.
- Yu, F.; Huang, H.; Yu, Q.; Ma, Y.; Zhang, Q.; Zhang, B. Artificial intelligence-based myocardial texture analysis in etiological differentiation of left ventricular hypertrophy. Ann. Transl. Med. 2021, 9, 108.
- Goto, S.; Mahara, K.; Beussink-Nelson, L.; Ikura, H.; Katsumata, Y.; Endo, J.; Gaggin, H.K.; Shah, S.J.; Itabashi, Y.; MacRae, C.A.; et al. Artificial intelligence-enabled fully automated detection of cardiac amyloidosis using electrocardiograms and echocardiograms. Nat. Commun. 2021, 12.
- Augusto, J.B.; Davies, R.H.; Bhuva, A.N.; Knott, K.D.; Seraphim, A.; Alfarih, M.; Lau, C.; Hughes, R.K.; Lopes, L.R.; Shiwani, H.; et al. Diagnosis and risk stratification in hypertrophic cardiomyopathy using machine learning wall thickness measurement: A comparison with human test-retest performance. Lancet Digit. Health 2021, 3, e20–e28.
- Zhang, J.; Deo, R.C. Response by Zhang and Deo to Letter Regarding Article, “Fully Automated Echocardiogram Interpretation in Clinical Practice: Feasibility and Diagnostic Accuracy”. Circulation 2019, 139, 1648–1649.
- Sado, D.M.; White, S.K.; Piechnik, S.K.; Banypersad, S.M.; Treibel, T.; Captur, G.; Fontana, M.; Maestrini, V.; Flett, A.S.; Robson, M.D.; et al. Identification and assessment of anderson-fabry disease by cardiovascular magnetic resonance noncontrast myocardial T1 mapping. Circ. Cardiovasc. Imaging 2013, 6, 392–398.
- Pica, S.; Sado, D.M.; Maestrini, V.; Fontana, M.; White, S.K.; Treibel, T.; Captur, G.; Anderson, S.; Piechnik, S.K.; Robson, M.D.; et al. Reproducibility of native myocardial T1 mapping in the assessment of Fabry disease and its role in early detection of cardiac involvement by cardiovascular magnetic resonance. J. Cardiovasc. Magn. Reson. 2014, 16, 99.
- Augusto, J.B.; Nordin, S.; Vijapurapu, R.; Baig, S.; Bulluck, H.; Castelletti, S.; Alfarih, M.; Knott, K.; Captur, G.; Kotecha, T.; et al. Myocardial edema, myocyte injury, and disease severity in Fabry disease. Circ. Cardiovasc. Imaging 2020, 13, 10171.
- Seydelmann, N.; Liu, D.; Krämer, J.; Drechsler, C.; Hu, K.; Nordbeck, P.; Schneider, A.; Störk, S.; Bijnens, B.; Ertl, G.; et al. High-sensitivity troponin: A clinical blood biomarker for staging cardiomyopathy in fabry disease. J. Am. Heart Assoc. 2016, 5.
- Yogasundaram, H.; Nikhanj, A.; Putko, B.N.; Boutin, M.; Jain-Ghai, S.; Khan, A.; Auray-Blais, C.; West, M.L.; Oudit, G.Y. Elevated inflammatory plasma biomarkers in patients with fabry disease: A critical link to heart failure with preserved ejection fraction. J. Am. Heart Assoc. 2018, 7.
- Altarescu, G.; Chicco, G.; Whybra, C.; Delgado-Sanchez, S.; Sharon, N.; Beck, M.; Elstein, D. Correlation between interleukin-6 promoter and C-reactive protein (CRP) polymorphisms and CRP levels with the Mainz Severity Score Index for Fabry disease. J. Inherit. Metab. Dis. 2008, 31, 117–123.
- Coats, C.J.; Parisi, V.; Ramos, M.; Janagarajan, K.; O’Mahony, C.; Dawnay, A.; Lachmann, R.H.; Murphy, E.; Mehta, A.; Hughes, D.; et al. Role of serum N-terminal pro-brain natriuretic peptide measurement in diagnosis of cardiac involvement in patients with anderson-fabry disease. Am. J. Cardiol. 2013, 111, 111–117.
- Lobo, T.; Morgan, J.; Bjorksten, A.; Nicholls, K.; Grigg, L.; Centra, E.; Becker, G. Cardiovascular testing in Fabry disease: Exercise capacity reduction, chronotropic incompetence and improved anaerobic threshold after enzyme replacement. Intern. Med. J. 2008, 38, 407–414.
- Bierer, G.; Kamangar, N.; Balte, D.; Wilcox, W.R.; Mosenifar, Z. Cardiopulmonary exercise testing in fabry disease. Respiration 2005, 72, 504–511.
- Powell, A.W.; Jefferies, J.L.; Hopkin, R.J.; Mays, W.A.; Goa, Z.; Chin, C. Cardiopulmonary fitness assessment on maximal and submaximal exercise testing in patients with Fabry disease. Am. J. Med. Genet. Part A 2018, 176, 1852–1857.
- Bierer, G.; Balfe, D.; Wilcox, W.R.; Mosenifar, Z. Improvement in serial cardiopulmonary exercise testing following enzyme replacement therapy in Fabry disease. J. Inherit. Metab. Dis. 2006, 29, 572–579.
- Khan, A.; Barber, D.L.; Huang, J.; Rupar, C.A.; Rip, J.W.; Auray-Blais, C.; Boutin, M.; O’Hoski, P.; Gargulak, K.; McKillop, W.M.; et al. Lentivirus-mediated gene therapy for Fabry disease. Nat. Commun. 2021, 12.
- Eng, C.M.; Guffon, N.; Wilcox, W.R.; Germain, D.P.; Lee, P.; Waldek, S.; Caplan, L.; Linthorst, G.E.; Desnick, R.J. Safety and Efficacy of Recombinant Human α-Galactosidase A Replacement Therapy in Fabry’s Disease. N. Engl. J. Med. 2001, 345, 9–16.
- Weidemann, F.; Niemann, M.; Breunig, F.; Herrmann, S.; Beer, M.; Störk, S.; Voelker, W.; Ertl, G.; Wanner, C.; Strotmann, J. Long-term effects of enzyme replacement therapy on fabry cardiomyopathy. Evidence for a better outcome with early treatment. Circulation 2009, 119, 524–529.
- Weidemann, F.; Breunig, F.; Beer, M.; Sandstede, J.; Turschner, O.; Voelker, W.; Ertl, G.; Knoll, A.; Wanner, C.; Strotmann, J.M. Improvement of cardiac function during enzyme replacement therapy in patients with fabry disease: A prospective strain rate imaging study. Circulation 2003, 108, 1299–1301.
- Van der Veen, S.J.; Hollak, C.E.M.; Van Kuilenburg, A.B.P.; Langeveld, M. Developments in the treatment of Fabry disease. J. Inherit. Metab. Dis. 2020, 43, 908–921.
- Schiffmann, R.; Kopp, J.B.; Austin, H.A.; Balow, J.E.; Brady, R.O. Enzyme Replacement Therapy in Fabry Disease: A Randomized Controlled Trial. N. Engl. J. Med. 2001, 285, 2743.
- El Dib, R.; Gomaa, H.; Carvalho, R.P.; Camargo, S.E.; Bazan, R.; Barretti, P.; Barreto, F.C. Enzyme replacement therapy for Anderson-Fabry disease. Cochrane Database Syst. Rev. 2016.
- Sirrs, S.M.; Bichet, D.G.; Casey, R.; Clarke, J.T.R.; Lemoine, K.; Doucette, S.; West, M.L. Outcomes of patients treated through the Canadian Fabry disease initiative. Mol. Genet. Metab. 2014, 111, 499–506.
- Arends, M.; Biegstraaten, M.; Wanner, C.; Sirrs, S.; Mehta, A.; Elliott, P.M.; Oder, D.; Watkinson, O.T.; Bichet, D.G.; Khan, A.; et al. Agalsidase alfa versus agalsidase beta for the treatment of Fabry disease: An international cohort study. J. Med. Genet. 2018, 55, 351–358.
- Germain, D.P.; Hughes, D.A.; Nicholls, K.; Bichet, D.G.; Giugliani, R.; Wilcox, W.R.; Feliciani, C.; Shankar, S.P.; Ezgu, F.; Amartino, H.; et al. Treatment of Fabry’s Disease with the Pharmacologic Chaperone Migalastat. N. Engl. J. Med. 2016, 375, 545–555.
- Germain, D.P.; Fan, J.Q. Pharmacological chaperone therapy by active-site-specific chaperones in Fabry disease: In vitro and preclinical studies. Int. J. Clin. Pharmacol. Ther. 2009, 47, S111–S117.
- Hughes, D.A.; Nicholls, K.; Shankar, S.P.; Sunder-Plassmann, G.; Koeller, D.; Nedd, K.; Vockley, G.; Hamazaki, T.; Lachmann, R.; Ohashi, T.; et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med. Genet. 2017, 54, 288–296.
- Germain, D.P.; Nicholls, K.; Giugliani, R.; Bichet, D.G.; Hughes, D.A.; Barisoni, L.M.; Colvin, R.B.; Jennette, J.C.; Skuban, N.; Castelli, J.P.; et al. Efficacy of the pharmacologic chaperone migalastat in a subset of male patients with the classic phenotype of Fabry disease and migalastat-amenable variants: Data from the phase 3 randomized, multicenter, double-blind clinical trial and extension study. Genet. Med. 2019, 21, 1987–1997.

