 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Giorgio Merlo | + 2989 word(s) | 2989 | 2021-06-16 11:55:36 | | | |
| 2 | Amina Yu | Meta information modification | 2989 | 2021-06-18 03:31:40 | | |
Video Upload Options
Intellectual disability (ID) is a condition characterized by limited intellectual functioning and adaptive behaviors. It affects 1–3% of the worldwide population and no pharmacological therapies are currently available. Too many genes have been found mutated in ID patients suggesting that the genetic bases are highly heterogeneous and apparently unrelated. Bibliomic analysis reveals that ID genes converge onto a few biological modules, one of these being cytoskeleton dynamics and Rho GTPases transduction. Genetic variants exert their effects at different levels in a hierarchical arrangement, starting from the molecular level and moving toward higher levels of organization, i.e., cell compartment and functions, circuits, cognition, and behavior. Thus, cytoskeleton alterations which have an impact on cell processes such as neuronal migration, neuritogenesis, and synaptic plasticity, rebound on the overall establishment of an effective network and consequently on the cognitive phenotype. Systems biology approaches are more focused on the overall interconnected network rather than on individual genes, encouraging the design of therapies that aim to correct common dysregulated biological processes. This review summarizes current knowledge about cytoskeleton control in neurons and its relevance for the ID pathogenesis, exploiting in silico modeling and translating the implications of those findings to biomedical research.
1. Introduction
2. Cytoskeleton Functions in Neuronal Development
2.1. The Core Regulation of Actin Dynamics
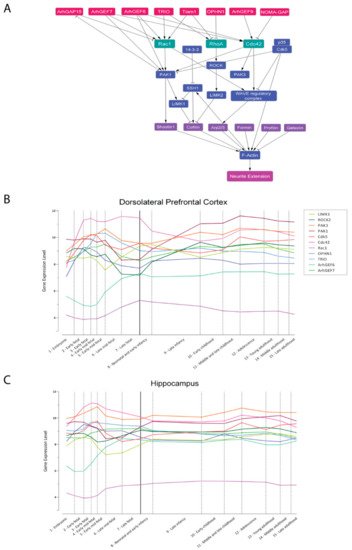
2.2. Synaptogenesis and Synaptic Plasticity
2.3. The Role of Microtubules in ID
3. Concluding Remarks
References
- Kochinke, K.; Zweier, C.; Nijhof, B.; Fenckova, M.; Cizek, P.; Honti, F.; Keerthikumar, S.; Oortveld, M.A.; Kleefstra, T.; Kramer, J.; et al. Systematic Phenomics Analysis Deconvolutes Genes Mutated in Intellectual Disability into Biologically Coherent Modules. Am. J. Hum. Genet. 2016, 98, 149–164.
- Mir, Y.R.; Kuchay, R.A.H. Advances in identification of genes involved in autosomal recessive intellectual disability: A brief review. J. Med. Genet. 2019, 56, 567–573.
- Van Bokhoven, H. Genetic and Epigenetic Networks in Intellectual Disabilities. Annu. Rev. Genet. 2011, 45, 81–104.
- Srivastava, A.K.; Schwartz, C.E. Intellectual disability and autism spectrum disorders: Causal genes and molecular mechanisms. Neurosci. Biobehav. Rev. 2014, 46, 161–174.
- McKenzie, K.; Milton, M.; Smith, G.; Ouellette-Kuntz, H. Systematic Review of the Prevalence and Incidence of Intellectual Disabilities: Current Trends and Issues. Curr. Dev. Disord. Rep. 2016, 3, 104–115.
- Mottron, L.; Belleville, S.; Rouleau, G.A.; Collignon, O. Linking neocortical, cognitive, and genetic variability in autism with alterations of brain plasticity: The Trigger-Threshold-Target model. Neurosci. Biobehav. Rev. 2014, 47, 735–752.
- Ba, W.; van der Raadt, J.; Kasri, N.N. Rho GTPase signaling at the synapse: Implications for intellectual disability. Exp. Cell Res. 2013, 319, 2368–2374.
- Penzes, P.; Buonanno, A.; Passafarro, M.; Sala, C.; Sweet, R.A. Developmental vulnerability of synapses and circuits associated with neuropsychiatric disorders. J. Neurochem. 2013, 126, 165–182.
- Murakoshi, H.; Wang, H.; Yasuda, R. Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nat. Cell Biol. 2011, 472, 100–104.
- Tejada-Simon, M.V. Modulation of actin dynamics by Rac1 to target cognitive function. J. Neurochem. 2015, 133, 767–779.
- De Filippis, B.; Valenti, D.; Chiodi, V.; Ferrante, A.; de Bari, L.; Fiorentini, C.; Domenici, M.R.; Ricceri, L.; Vacca, R.A.; Fabbri, A.; et al. Modulation of Rho GTPases rescues brain mitochondrial dysfunction, cognitive deficits and aberrant synaptic plasticity in female mice modeling Rett syndrome. Eur. Neuropsychopharmacol. 2015, 25, 889–901.
- Kahn, O.I.; Baas, P.W. Microtubules and Growth Cones: Motors Drive the Turn. Trends Neurosci. 2016, 39, 433–440.
- Lowery, L.A.; Van Vactor, D. The trip of the tip: Understanding the growth cone machinery. Nat. Rev. Mol. Cell Biol. 2009, 10, 332–343.
- Maday, S.; Twelvetrees, A.E.; Moughamian, A.J.; Holzbaur, E.L. Axonal Transport: Cargo-Specific Mechanisms of Motility and Regulation. Neuron 2014, 84, 292–309.
- Tanaka, E.; Sabry, J. Making the connection: Cytoskeletal rearrangements during growth cone guidance. Cell 1995, 83, 171–176.
- Lewis, S.A.; Tian, G.; Cowan, N.J. The α- and β-tubulin folding pathways. Trends Cell Biol. 1997, 7, 479–484.
- Kunze, D.; Rüstow, B. Pathobiochemical Aspects of Cytoskeleton Components. Clin. Chem. Lab. Med. 1993, 31, 477–490.
- Johnson, G.V.W.; Jope, R.S. The role of microtubule-associated protein 2 (MAP-2) in neuronal growth, plasticity, and degeneration. J. Neurosci. Res. 1992, 33, 505–512.
- Winder, S.J.; Ayscough, K.R. Actin-binding proteins. J. Cell Sci. 2005, 118, 651–654.
- Mitchison, T.J.; Kirschner, M.W. Dynamic instability of microtubule growth. Nat. Cell Biol. 1984, 312, 237–242.
- Pollard, T.D.; Borisy, G.G. Cellular Motility Driven by Assembly and Disassembly of Actin Filaments. Cell 2003, 112, 453–465.
- Haydon, P.G.; Drapeau, P. From contact to connection: Early events during synaptogenesis. Trends Neurosci. 1995, 18, 196–201.
- Korobova, F.; Svitkina, T. Molecular Architecture of Synaptic Actin Cytoskeleton in Hippocampal Neurons Reveals a Mechanism of Dendritic Spine Morphogenesis. Mol. Biol. Cell 2010, 21, 165–176.
- Südhof, T.C. Towards an Understanding of Synapse Formation. Neuron 2018, 100, 276–293.
- Yoshihara, Y.; De Roo, M.; Muller, D. Dendritic spine formation and stabilization. Curr. Opin. Neurobiol. 2009, 19, 146–153.
- Spence, E.F.; Soderling, S.H. Actin Out: Regulation of the Synaptic Cytoskeleton. J. Biol. Chem. 2015, 290, 28613–28622.
- Yusifov, R.; Tippmann, A.; Staiger, J.F.; Schlüter, O.M.; Löwel, S. Spine dynamics of PSD-95-deficient neurons in the visual cortex link silent synapses to structural cortical plasticity. Proc. Natl. Acad. Sci. USA 2021, 118, e2022701118.
- Sala, C.; Piëch, V.; Wilson, N.R.; Passafaro, M.; Liu, G.; Sheng, M. Regulation of Dendritic Spine Morphology and Synaptic Function by Shank and Homer. Neuron 2001, 31, 115–130.
- Repetto, D.; Camera, P.; Melani, R.; Morello, N.; Russo, I.; Calcagno, E.; Tomasoni, R.; Bianchi, F.T.; Berto, G.; Giustetto, M.; et al. P140cap Regulates Memory and Synaptic Plasticity through Src-Mediated and Citron-N-Mediated Actin Reorganization. J. Neurosci. 2014, 34, 1542–1553.
- Huganir, R.L.; Nicoll, R.A. AMPARs and Synaptic Plasticity: The Last 25 Years. Neuron 2013, 80, 704–717.
- Allison, D.W.; Gelfand, V.I.; Spector, I.; Craig, A.M. Role of Actin in Anchoring Postsynaptic Receptors in Cultured Hippocampal Neurons: Differential Attachment of NMDA versus AMPA Receptors. J. Neurosci. 1998, 18, 2423–2436.
- Kim, C.-H.; Lisman, J.E. A Role of Actin Filament in Synaptic Transmission and Long-Term Potentiation. J. Neurosci. 1999, 19, 4314–4324.
- Zhou, Q.; Xiao, M.-Y.; Nicoll, R.A. Contribution of cytoskeleton to the internalization of AMPA receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 1261–1266.
- Duffney, L.J.; Wei, J.; Cheng, J.; Liu, W.; Smith, K.R.; Kittler, J.T.; Yan, Z. Shank3 Deficiency Induces NMDA Receptor Hypofunction via an Actin-Dependent Mechanism. J. Neurosci. 2013, 33, 15767–15778.
- Irwin, S.A.; Galvez, R.; Greenough, W.T. Dendritic Spine Structural Anomalies in Fragile-X Mental Retardation Syndrome. Cereb. Cortex 2000, 10, 1038–1044.
- Kozma, R.; Sarner, S.; Ahmed, S.; Lim, L. Rho family GTPases and neuronal growth cone remodelling: Relationship between increased complexity induced by Cdc42Hs, Rac1, and acetylcholine and collapse induced by RhoA and lysophosphatidic acid. Mol. Cell. Biol. 1997, 17, 1201–1211.
- Smith, C.L. Cytoskeletal movements and substrate interactions during initiation of neurite outgrowth by sympathetic neurons in vitro. J. Neurosci. 1994, 14, 384–398.
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504.
- Kang, H.J.; Kawasawa, Y.I.; Cheng, F.; Zhu, Y.; Xu, X.; Li, M.; Sousa, A.M.M.; Pletikos, M.; Meyer, K.A.; Sedmak, G.; et al. Spatio-temporal transcriptome of the human brain. Nature 2011, 478, 483–489.
- Govek, E.E.; Newey, S.E.; Van Aelst, L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005, 19, 1–49.
- Ma, X.M.; Huang, J.; Wang, Y.; Eipper, B.A.; Mains, R.E. Kalirin, a Multifunctional Rho Guanine Nucleotide Exchange Factor, Is Necessary for Maintenance of Hippocampal Pyramidal Neuron Dendrites and Dendritic Spines. J. Neurosci. 2003, 23, 10593–10603.
- Ma, X.M.; Kiraly, D.D.; Gaier, E.D.; Wang, Y.; Kim, E.J.; Levine, E.S.; Eipper, B.A.; Mains, R.E. Kalirin-7 is required for synaptic structure and function. J. Neurosci. 2008, 28, 12368–12382.
- Satoh, A.; Nakanishi, H.; Obaishi, H.; Wada, M.; Takahashi, K.; Satoh, K.; Hirao, K.; Nishioka, H.; Hata, Y.; Mizoguchi, A.; et al. Neurabin-II/spinophilin: An actin filament-binding protein with one PDZ domain localized at cadherin-based cell-cell adhesion sites. J. Biol. Chem. 1998, 273, 3470–3475.
- Grossman, S.D.; Hsieh-Wilson, L.C.; Allen, P.B.; Nairn, A.C.; Greengard, P. The actin-binding domain of spinophilin is necessary and sufficient for targeting to dendritic spines. Neuro Mol. Med. 2002, 2, 61–69.
- Feng, J.; Yan, Z.; Ferreira, A.; Tomizawa, K.; Liauw, J.A.; Zhuo, M.; Allen, P.B.; Ouimet, C.C.; Greengard, P. Spinophilin regulates the formation and function of dendritic spines. Proc. Natl. Acad. Sci. USA 2000, 97, 9287–9292.
- Ryan, X.P.; Alldritt, J.; Svenningsson, P.; Allen, P.B.; Wu, G.Y.; Nairn, A.C.; Greengard, P. The Rho-specific GEF Lfc interacts with neurabin and spinophilin to regulate dendritic spine morphology. Neuron 2005, 47, 85–100.
- Ravindran, E.; Hu, H.; Yuzwa, S.A.; Hernandez-Miranda, L.R.; Kraemer, N.; Ninnemann, O.; Musante, L.; Boltshauser, E.; Schindler, D.; Hübner, A.; et al. Homozygous ARHGEF2 mutation causes intellectual disability and midbrain-hindbrain malformation. PLoS Genet. 2017, 13, e1006746.
- Qualmann, B.; Boeckers, T.M.; Jeromin, M.; Gundelfinger, E.D.; Kessels, M.M. Linkage of the Actin Cytoskeleton to the Postsynaptic Density via Direct Interactions of Abp1 with the ProSAP/Shank Family. J. Neurosci. 2004, 24, 2481–2495.
- Park, E.; Na, M.; Choi, J.; Kim, S.; Lee, J.R.; Yoon, J.; Park, D.; Sheng, M.; Kim, E. The Shank family of postsynaptic density proteins interacts with and promotes synaptic accumulation of the βPIX guanine nucleotide exchange factor for Rac1 and Cdc42. J. Biol. Chem. 2003, 278, 19220–19229.
- Duffney, L.J.; Zhong, P.; Wei, J.; Matas, E.; Cheng, J.; Qin, L.; Ma, K.; Dietz, D.M.; Kajiwara, Y.; Buxbaum, J.D.; et al. Autism-like Deficits in Shank3-Deficient Mice Are Rescued by Targeting Actin Regulators. Cell Rep. 2015, 11, 1400–1413.
- Han, K.; Holder, J.L.; Schaaf, C.P.; Lu, H.; Chen, H.; Kang, H.; Tang, J.; Wu, Z.; Hao, S.; Cheung, S.W.; et al. SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature 2013, 503, 72–77.
- Sarowar, T.; Grabrucker, A.M. Actin-Dependent Alterations of Dendritic Spine Morphology in Shankopathies. Neural Plast. 2016, 2016, 8051861.
- Bonaglia, M.C.; Giorda, R.; Mani, E.; Aceti, G.; Anderlid, B.M.; Baroncini, A.; Pramparo, T.; Zuffardi, O. Identification of a recurrent breakpoint within the SHANK3 gene in the 22q13.3 deletion syndrome. J. Med. Genet. 2006, 43, 822–828.
- Bonaglia, M.C.; Giorda, R.; Borgatti, R.; Felisari, G.; Gagliardi, C.; Selicorni, A.; Zuffardi, O. Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. Am. J. Hum. Genet. 2001, 69, 261–268.
- Soorya, L.; Kolevzon, A.; Zweifach, J.; Lim, T.; Dobry, Y.; Schwartz, L.; Frank, Y.; Wang, A.T.; Cai, G.; Parkhomenko, E.; et al. Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and SHANK3 deficiency. Mol. Autism 2013, 4, 18.
- Holder, J.L., Jr.; Hamdan, F.F.; Michaud, J.L. SYNGAP1-Related Intellectual Disability; University of Washington: Seattle, WA, USA, 1993.
- Shen, K.; Teruel, M.N.; Subramanian, K.; Meyer, T. CaMKIIβ functions as an F-actin targeting module that localizes CaMKIIα/β heterooligomers to dendritic spines. Neuron 1998, 21, 593–606.
- Küry, S.; van Woerden, G.M.; Besnard, T.; Proietti Onori, M.; Latypova, X.; Towne, M.C.; Cho, M.T.; Prescott, T.E.; Ploeg, M.A.; Sanders, S.; et al. De Novo Mutations in Protein Kinase Genes CAMK2A and CAMK2B Cause Intellectual Disability. Am. J. Hum. Genet. 2017, 101, 768–788.
- Lasser, M.; Tiber, J.; Lowery, L.A. The role of the microtubule cytoskeleton in neurodevelopmental disorders. Front. Cell. Neurosci. 2018, 12, 165.
- Conde, C.; Cáceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci. 2009, 10, 319–332.
- Witte, H.; Neukirchen, D.; Bradke, F. Microtubule stabilization specifies initial neuronal polarization. J. Cell Biol. 2008, 180, 619–632.
- Gu, J.; Firestein, B.L.; Zheng, J.Q. Microtubules in dendritic spine development. J. Neurosci. 2008, 28, 12120–12124.
- Jaworski, J.; Kapitein, L.C.; Gouveia, S.M.; Dortland, B.R.; Wulf, P.S.; Grigoriev, I.; Camera, P.; Spangler, S.A.; Di Stefano, P.; Demmers, J.; et al. Dynamic Microtubules Regulate Dendritic Spine Morphology and Synaptic Plasticity. Neuron 2009, 61, 85–100.
- Dent, E.W. Of microtubules and memory: Implications for microtubule dynamics in dendrites and spines. Mol. Biol. Cell 2017, 28, 1–8.
- Karabay, A.; Yu, W.; Solowska, J.M.; Baird, D.H.; Baas, P.W. Axonal growth is sensitive to the levels of katanin, a protein that severs microtubules. J. Neurosci. 2004, 24, 5778–5788.
- Akhmanova, A.; Steinmetz, M.O. Tracking the ends: A dynamic protein network controls the fate of microtubule tips. Nat. Rev. Mol. Cell Biol. 2008, 9, 309–322.
- Jaworski, J.; Hoogenraad, C.C.; Akhmanova, A. Microtubule plus-end tracking proteins in differentiated mammalian cells. Int. J. Biochem. Cell Biol. 2008, 40, 619–637.
- Neukirchen, D.; Bradke, F. Cytoplasmic linker proteins regulate neuronal polarization through microtubule and growth cone dynamics. J. Neurosci. 2011, 31, 1528–1538.
- Swiech, L.; Blazejczyk, M.; Urbanska, M.; Pietruszka, P.; Dortland, B.R.; Malik, A.R.; Wulf, P.S.; Hoogenraad, C.C.; Jaworski, J. CLIP-170 and IQGAP1 cooperatively regulate dendrite morphology. J. Neurosci. 2011, 31, 4555–4568.
- Yonekawa, V.; Harada, A.; Okada, Y.; Funakoshi, T.; Kanai, Y.; Takei, Y.; Terada, S.; Noda, T.; Hirokawa, N. Defect in synaptic vesicle precursor transport and neuronal cell death in KIF1A motor protein-deficient mice. J. Cell Biol. 1998, 141, 431–441.
- Hamdan, F.F.; Gauthier, J.; Araki, Y.; Lin, D.T.; Yoshizawa, Y.; Higashi, K.; Park, A.R.; Spiegelman, D.; Dobrzeniecka, S.; Piton, A.; et al. Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am. J. Hum. Genet. 2011, 88, 306–316.
- Esmaeeli Nieh, S.; Madou, M.R.Z.; Sirajuddin, M.; Fregeau, B.; Mcknight, D.; Lexa, K.; Strober, J.; Spaeth, C.; Hallinan, B.E.; Smaoui, N.; et al. De novo mutations in KIF1A cause progressive encephalopathy and brain atrophy. Ann. Clin. Transl. Neurol. 2015, 2, 623–635.
- Willemsen, M.H.; Ba, W.; Wissink-Lindhout, W.M.; de Brouwer, A.P.M.; Haas, S.A.; Bienek, M.; Hu, H.; Vissers, L.E.L.M.; van Bokhoven, H.; Kalscheuer, V.; et al. Involvement of the kinesin family members KIF4A and KIF5C in intellectual disability and synaptic function. J. Med. Genet. 2014, 51, 487–494.
- Poirier, K.; Lebrun, N.; Broix, L.; Tian, G.; Saillour, Y.; Boscheron, C.; Parrini, E.; Valence, S.; Saint Pierre, B.; Oger, M.; et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat. Genet. 2013, 45, 639–647.
- Putoux, A.; Thomas, S.; Coene, K.L.M.; Davis, E.E.; Alanay, Y.; Ogur, G.; Uz, E.; Buzas, D.; Gomes, C.; Patrier, S.; et al. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat. Genet. 2011, 43, 601–606.
- Kevenaar, J.T.; Bianchi, S.; Van Spronsen, M.; Olieric, N.; Lipka, J.; Frias, C.P.; Mikhaylova, M.; Harterink, M.; Keijzer, N.; Wulf, P.S.; et al. Kinesin-Binding Protein Controls Microtubule Dynamics and Cargo Trafficking by Regulating Kinesin Motor Activity. Curr. Biol. 2016, 26, 849–861.
- Hempel, M.; Cremer, K.; Ockeloen, C.W.; Lichtenbelt, K.D.; Herkert, J.C.; Denecke, J.; Haack, T.B.; Zink, A.M.; Becker, J.; Wohlleber, E.; et al. De Novo Mutations in CHAMP1 Cause Intellectual Disability with Severe Speech Impairment. Am. J. Hum. Genet. 2015, 97, 493–500.
- Isidor, B.; Küry, S.; Rosenfeld, J.A.; Besnard, T.; Schmitt, S.; Joss, S.; Davies, S.J.; Roger Lebel, R.; Henderson, A.; Schaaf, C.P.; et al. De Novo Truncating Mutations in the Kinetochore-Microtubules Attachment Gene CHAMP1 Cause Syndromic Intellectual Disability. Hum. Mutat. 2016, 37, 354–358.
- Tanaka, A.J.; Cho, M.T.; Retterer, K.; Jones, J.R.; Nowak, C.; Douglas, J.; Jiang, Y.-H.; McConkie-Rosell, A.; Schaefer, G.B.; Kaylor, J.; et al. De novo pathogenic variants in CHAMP1 are associated with global developmental delay, intellectual disability, and dysmorphic facial features. Mol. Case Stud. 2016, 2, a000661.
- Larti, F.; Kahrizi, K.; Musante, L.; Hu, H.; Papari, E.; Fattahi, Z.; Bazazzadegan, N.; Liu, Z.; Banan, M.; Garshasbi, M.; et al. Erratum: A defect in the CLIP1 gene (CLIP-170) can cause autosomal recessive intellectual disability. Eur. J. Hum. Genet. 2015, 23, 416.
- Bartholdi, D.; Stray-Pedersen, A.; Azzarello-Burri, S.; Kibaek, M.; Kirchhoff, M.; Oneda, B.; Rødningen, O.; Schmitt-Mechelke, T.; Rauch, A.; Kjaergaard, S. A newly recognized 13q12.3 microdeletion syndrome characterized by intellectual disability, microcephaly, and eczema/atopic dermatitis encompassing the HMGB1 and KATNAL1 genes. Am. J. Med. Genet. Part A 2014, 164, 1277–1283.
- Cainarca, S.; Messali, S.; Ballabio, A.; Meroni, G. Functional characterization of the Opitz syndrome gene product (midin): Evidence for homodimerization and association with microtubules throughout the cell cycle. Hum. Mol. Genet. 1999, 8, 1387–1396.
- Geetha, T.S.; Michealraj, K.A.; Kabra, M.; Kaur, G.; Juyal, R.C.; Thelma, B.K. Targeted Deep Resequencing Identifies MID2 Mutation for X-Linked Intellectual Disability with Varied Disease Severity in a Large Kindred from India. Hum. Mutat. 2014, 35, 41–44.
- Barbiero, I.; Peroni, D.; Tramarin, M.; Chandola, C.; Rusconi, L.; Landsberger, N.; Kilstrup-Nielsen, C. The neurosteroid pregnenolone reverts microtubule derangement induced by the loss of a functional CDKL5-IQGAP1 complex. Hum. Mol. Genet. 2017, 26, 3520–3530.
- Baltussen, L.L.; Negraes, P.D.; Silvestre, M.; Claxton, S.; Moeskops, M.; Christodoulou, E.; Flynn, H.R.; Snijders, A.P.; Muotri, A.R.; Ultanir, S.K. Chemical genetic identification of CDKL5 substrates reveals its role in neuronal microtubule dynamics. EMBO J. 2018, 37, e99763.
- Nawaz, M.S.; Giarda, E.; Bedogni, F.; La Montanara, P.; Ricciardi, S.; Ciceri, D.; Alberio, T.; Landsberger, N.; Rusconi, L.; Kilstrup-Nielsen, C. CDKL5 and shootin1 interact and concur in regulating neuronal polarization. PLoS ONE 2016, 11, e0148634.
- Fehr, S.; Wilson, M.; Downs, J.; Williams, S.; Murgia, A.; Sartori, S.; Vecchi, M.; Ho, G.; Polli, R.; Psoni, S.; et al. The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur. J. Hum. Genet. 2013, 21, 266–273.
- Guy, J.; Gan, J.; Selfridge, J.; Cobb, S.; Bird, A. Reversal of neurological defects in a mouse model of Rett syndrome. Science 2007, 315, 1143–1147.
- Corti, S.; Faravelli, I.; Cardano, M.; Conti, L. Human pluripotent stem cells as tools for neurodegenerative and neurodevelopmental disease modeling and drug discovery. Expert Opin. Drug Discov. 2015, 10, 615–629.

