 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jacek Szymański | + 2416 word(s) | 2416 | 2021-05-11 07:50:23 | | | |
| 2 | Nora Tang | Meta information modification | 2416 | 2021-06-11 04:56:19 | | |
Video Upload Options
The aging of the organism is a complex and multifactorial process. It can be viewed in the context of the whole organism, but also of individual tissues and organs. The problem of vaginal aging and the related genitourinary syndrome of menopause significantly reduces the quality of women’s lives. The aging process of the vagina includes estrogen deficiencies, changes in the microbiome, and changes at the genetic level associated with DNA methylation. During the menopause, the number of Lactobacillus colonies decreases, and the number of pathological bacteria colonies increases. The decrease in estrogen levels results in a decrease in vaginal epithelial permeability, perfusion, and elastin levels, resulting in vaginal dryness and atrophy. Changes at the molecular level are the least clear. It can also be assumed that, similarly to the tissues studied so far, there are changes in cytosine methylation and TET (ten-eleven translocation) expression. The interrelationships between DNA methylation, hormonal changes, and the vaginal microbiome have not yet been fully elucidated.
1. Introduction
The aging of the organism is an extremely complex and multifactorial process. It consists of changes at the genetic, hormonal, and immunological levels. Moreover, the problem of aging can be considered in terms of the whole organism, individual organs, and tissues. The aim of this study is to present the current knowledge on the aging of the vagina. So far, the vaginal microbiota and the effects of estrogens on the vaginal epithelium, as well as the effects of estrogen deficiency, have been well understood. Undoubtedly, the vaginal microbiome is dependent on estrogen levels. However, there are no studies on the genetic aspects of the aging of the vaginal mucosa. It has been proven that DNA (deoxyribonucleic acid) methylation is associated with the aging process [1][2]. However, it is unclear whether DNA methylation is the cause of aging, an effect of it, or just a concomitant process. The analysis of DNA methylation in other tissues confirms the tissue specificity of this process. It can be presumed that the processes of DNA methylation and demethylation in the vagina will also have a specific course, and that the molecular processes of vaginal aging involve the cycle of dependence with the influence of estrogens and the microbiome. However, certain relationships have not yet been clarified (see Figure 1).
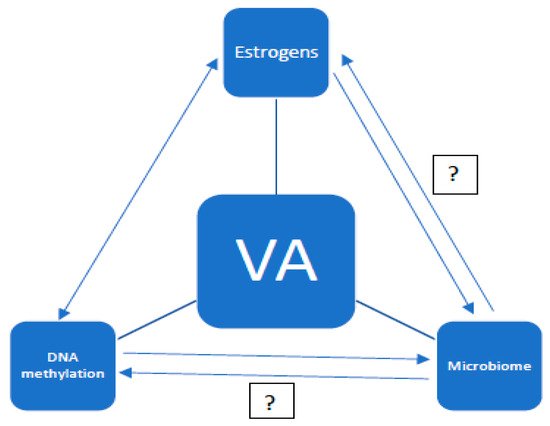
Figure 1. Potential relationships between estrogen levels, the vaginal microbiome, and DNA methylation in vaginal aging (VA—vaginal aging).
2. Changes in the Field of Microbiota
From the beginning of the existence of the human species, various bacterial colonies, generally known as the microbiota, inhabited different locations of the human body, and evolved along with the evolution of man. This co-evolution, as well as the transmission of microbes within a species over successive generations, has been proven on the basis of the convergence of the phylogenetic tree of microbiota and primates [3]. At the same time, an immune tolerance was developed, aimed at the elimination of invasive microbes and the protection of its own bacteria [4]. Socioeconomic development has caused changes in lifestyle, diet, and hygiene habits that, together with the introduction of antibiotics, has resulted in significant changes in the human microbiome. Another significant factor is the increase in deliveries by caesarean section in recent years, which disrupts the transmission of normal bacterial flora from the mother to the newborn. A microbiota disorder is currently believed to be the basis of many autoimmune diseases, allergies, obesity, and type II diabetes [5][6]. The gut microbiome has been best understood thus far. Changes in the intestinal microflora accompanying the aging of the organism have been observed. Moreover, a specific type of gut microbiota has been found in centenarians [7][8][9]. Another well-known microbiome is the vaginal flora dominated by the Lactobacillus species [10]. The Lactobacillus species are involved in the production of lactic acid and hydrogen peroxide, which ensure the right pH in the vagina, thus contributing to the protection of the vaginal environment against invasive bacterial strains [11]. A reduction in the number of Lactobacillus sp. often results in infection with anaerobic bacteria, leading to bacterial vaginosis and, in pregnant women, to preterm labor. The bacterial flora of the vagina dynamically changes under the influence of many factors, including lifestyle, smoking, hygiene habits, sex life, menstrual cycle, hormonal contraceptives, HPV (Human Papillomavirus) infections, Trichomonas vaginalis, and Chlamydia trachomatis, but mainly due to changes in the hormonal status of women resulting from age and the aging process [12][13][14][15][16][17][18][19]. The first significant changes in the bacterial environment of the vagina occur during puberty. Early puberty is characterized by a low Lactobacillus vaginal flora with the dominance of bacterial strains of intestinal origin. About 1 year before the menarche, the vaginal bacterial environment begins to resemble the microbiome of Lactobacillus-dominant women in their reproductive years [20]. The period of pregnancy is particularly important for the maintenance of normal bacterial flora, as the relationship between microbiota disorders and preterm labor has been proven [21][22]. During the menopause, a marked decrease in the number of Lactobacillus and an increase in colonies of pathological bacteria is observed, especially in women complaining of GSM (genitourinary syndrome of menopause) [11][23]. One study using the community state types (CSTs) scale showed marked differences in vaginal microbiomes between premenopausal, perimenopausal, and postmenopausal women. In women of reproductive age, CST-I or CST-III dominated; in perimenopausal CST-IVA, Lactobacillus gasseri dominated, while postmenopausal women were classified as CST-IVA/IVB or CSTV [13]. There was also a clear relationship between the bacterial state of the CST-IVA vagina and vulvovaginal atrophy [24]. What is more, hormone replacement therapy reducing GSM symptoms has a positive effect on the vaginal microbiome, restoring the dominance of Lactobacillus [25][26]. It can therefore be hypothesized that bacterial flora, dominated by Lactobacillus in postmenopausal women, improves quality of life.
3. Genetic Aspects of Aging
It is known that the ageing process of the organism is influenced by epigenetic changes, such as DNA methylation, the modification of histone proteins, or the expression of non-coding RNA (ribonucleic acid). While aging is associated with disturbances in DNA methylation, it is unclear whether it is the changes in DNA methylation alone that contribute to the aging process. In general, global hypomethylation and local hypermethylation are observed in the aging process [1]. The methylation of cytosine in CpG (5′-C-phosphate-G-3′) pairs, densely packed around the promoter, inactivates the gene, while methylation of rare CpG pairs causes the activation of the gene. In turn, the demethylation of cytosine in DNA usually activates genes [27]. In a study by Borkowska and colleagues, an increase in 5 mC (5 methylcytosine) in subcutaneous adipose tissue was shown, and ASCs (Adipose Stem Cells) correlated with the aging of the organism, with simultaneous hypomethylation in differentially hydroxymethylated regions (DHMRs). There was no correlation between the global 5 mC level and the expression of any of the TET (ten-eleven translocation) genes, although an age-related increase in TET2 mRNA and TET3 proteins was observed. It is possible that the ability of TET to convert 5 mC to 5fC (5 fluorocytosine) decreases with age, leading to the accumulation of 5 mC [2]. Similarly, an increase in 5 mC was observed in senescent human T cells. A reduced expression of TET 1 and TET3 mRNA was also noted, as well as no change in the expression of TET2 mRNA. There was also a correlation between TET3 expression and 5 mC content, with no such correlation in TET 1 and TET2 [28].
In the aging process, there is a global decline in DNA methylation. At the same time, the overall content of the epigenetic marker 5 mC increases with age, with no increase in TET 1-3 mRNA expression [29]. In mammals, DNA methylation is mainly located in CpG dinucleotides. The distribution of the CpG varies. High-density CpGs, the so-called CpG islands (CGIs), most often unmethylated, are located within the promoter regions. Conversely, the regions of low CpG density located primarily in repetitive DNA are heavily methylated. The third group comprises the low methylated regions (LMRs), dependent on the transcription factor (TF) binding site [1].
DNA methylation plays an essential role in the regulation of gene expression in mammals. The methylation process is tissue- and cell-specific through the action of cytosine DNA methyltransferases (in mammals: DNMT1, DNMT3A, and DNMT3B) [1]. 5 mC, also known as the sixth base, plays a special role in the regulation of gene expression. The level of 5 mC in gene bodies positively correlates with gene expression [29]. With the aging of the organism, an increase or decrease in cytosine methylation is observed, depending on the type and region of DNA [28]. In the cerebellum, a genomic increase of 5 mC was noted, with no changes in its content in mitochondrial DNA. A similar increase in genomic 5 mC was observed in sperm [28]. The DNA methylation process generally silences genes, although the role of methylation in gene activation remains unclear. The influence of environmental factors on the methylation process and the related gene activation or silencing mechanism seems to be highly probable [1]. The correct DNA methylation/demethylation process regulating correct transcription in the cell requires an appropriate interaction of DNA methyltransferases and TET proteins [30]. DNA demethylation, opposing methylation, may be passive during cell division in the absence of methylation or active, replication-independent, with the participation of TET family methylcytosine dioxygenases (ten-eleven translocation) [31]. TET causes the oxidation of 5 mC, generated by cytosine methylation and catalyzed by DNA methyltransferases (DNMTs), to 5 mC, 5fC, and 5caC (5 carboxylcytosine) in DNA. Additionally, thymine DNA glycosylase (TDG) restores unmethylated cytosine through active demethylation. Active demethylation also includes the DNA repair mechanism involving the excision of methylated cytosine and its replacement with unmethylated cytosine—the base excision repair (BER) pathway [27][32]. Passive demethylation, or rather the prevention of passive DNA methylation, is related to the inability to recognize 5 mC by DNMT1. In this way, the TET family controls the 5 mC and 5 hmC (5 hydroxymethylcytosine) levels through active demethylation or the negative regulation of passive DNA methylation [29].
The family of ten-eleven translocation (TET) dioxygenases is made up of TET 1, TET2, and TET3. Proteins have a similar structure built by the amino-terminal CXXC zinc-finger domain and the carboxyl-terminal catalytic dioxygenase domain. The CXXC zinc-finger domain consists of approximately 60 amino acids and is only present in TET 1 and TET3. The TET2 CXXC domain is a separate gene called IDAX or CXXC4 that negatively regulates TET2 [29][33]. The catalytic domain is made up of a Cys-rich segment, a double-stranded beta helix (DSBH) domain, and a non-conserved low-complexity insert (NCLC). The CXXC zinc-finger domain is responsible for the attachment to unmethylated CpG dinucleotides in DNA. TET2, in turn, binds to CpG dinucleotides with a preference for 5 mC. The level of 5 hmC is in the range of 5–40% 5 mC, depending on the type of cell. Interestingly, DNMT3A and DNMT3B also have the ability to directly convert 5 mC to cytosine. Moreover, the TET family, apart from its direct influence on DNA, modifies histones, thus influencing their stability and interaction with other proteins. This process occurs by recruiting O-linked β-d-N-acetylglucosamine transferase (OGT) to chromatin by binding TET to VprBP. OGT is present in the cytoplasm and cell nuclei, catalyzes serine and threonine glycosylation, and regulates TET phosphorylation [29]. Three mechanisms of regulation of gene transcription have been described:
-
Chromatin remodeling regulated by histone GlcNAcylation;
-
Transcriptional activation induced by the GlcNAcylation of HCF1 (host cell factor 1) promoted by the interaction of TET2/3 with OGT;
-
Facilitation of the biding of transcriptional factor-like molecules to the promoter of associated genes by the OGT–TET3 complex [29].
By attaching to CpG-rich regions, TET inhibits DNA methyltransferase activity, while converting 5 mC to 5 hmC. TET catalytic activity requires the presence of three cofactors: 2-oxoglutarate, Fe(II), and ascorbic acid, which prevents the oxidation of Fe(II). Vitamin C, as a cofactor of TET enzymes, induces TET-dependent DNA demethylation [29]. TET gene expression varies from tissue to tissue. In general, TET1 and TET2 mRNA levels decrease while TET3 mRNA levels increase during cell differentiation. In addition, the regulation of TET proteins also takes place at a non-translational level through caspase-dependent degradation and calpains proteases. OGT, by the acetylation of TET3, can prevent the formation of TET3-catalyzed 5 mC. The TET family plays an essential role in the development and differentiation of stem cells, thus contributing to the development of the embryo at various stages. It is not yet clear how TETs are involved in gene regulation in different cell types [34].
Differentially methylated regions (DMRs) are believed to be involved in the regulation of gene transcription. The distribution of DMRs is tissue-specific [2]. Together with CpG, they create the so-called epigenetic clock, determining the degree of tissue aging and lifespan [1].
CpGs and hypermethylated DMRs are located primarily on the genes involved in cell differentiation and development, and particularly within bivalent chromatin domain promoters. This is the region where the Polycomb target genes are located that regulate the silencing gene and genes that control development. The TET1 complex with the Polycomb Repressive Complex2 (PRC2) participates in epigenetic plasticity in the process of cell differentiation. Cell differentiation is thus regulated by the interaction of DNA methylation/demethylation and histone methylation/demethylation [29]. So far, it is not clear whether methylation can be directly related to the degree of aging of tissues and whether methylation causes aging, or is the result of this process [1]. Recent reports link DNA methylation with an influence on the energy metabolism through the regulation of the C/EBP-beta transcription factor (CCAAT/enhancer-binding protein-beta). C/EBPs play a key role in regulating glucose and fat metabolism. Impaired demethylation of C/EBP-beta sites accelerates the aging process. C/EPB belongs to the superfamily of transcription factors—the basic leucine zipper (bZIP)—controlled by the mechanistic/mammalian target of the rapamycin complex 1 (mTORC1), considered to be one of the key regulators of lifespan and health. mTORC1, by regulating C/EBP expression, influences the activation of the metabolic processes that are characteristic of caloric restrictions, as well as the production of specific proteins, thus affecting life expectancy [1]. The associations between metabolic processes, postmenopausal hormonal changes in a woman’s life, and DNA methylation require clarification. The relationship between estrogen deficiency and DNA methylation seems to be of particular interest. Ulrich and colleagues have shown inverse associations between sex hormones in overweight postmenopausal women with low serum folate levels and DNA methylation. Interestingly, such correlation was not observed among women with high folate levels and those who used multivitamins. This negative association between DNA methylation and sex hormones was apparent in women with a lower level of methyl donor S-adenosylmethionine resulting from a lower one-carbon status. Thus, the authors formulated the hypothesis that one-carbon status may be an important factor in the relationship between sex hormones and DNA methylation. However, direct evidence linking hormonal status to the methylation level is lacking [35].
References
- Niehrs, C.; Calkhoven, C.F. Emerging Role of C/EBPβ and Epigenetic DNA Methylation in Ageing. Trends Genet. 2020, 36, 71–80.
- Borkowska, J.; Domaszewska-Szostek, A.; Kołodziej, P.; Wicik, Z.; Połosak, J.; Buyanovskaya, O.; Charzewski, L.; Stańczyk, M.; Noszczyk, B.; Puzianowska-Kuznicka, M. Alterations in 5 hmC level and genomic distribution in aging-related epigenetic drift in human adipose stem cells. Epigenomics 2020, 12, 423–437.
- Santoro, A.; Zhao, J.; Wu, L.; Carru, C.; Biagi, E.; Franceschi, C. Microbiomes other than the gut: Inflammaging and age-related diseases. Semin. Immunopathol. 2020, 42, 589–605.
- Dominguez-Bello, M.G.; Godoy-Vitorino, F.; Knight, R. Role of the microbiome in human development. Gut 2019, 68, 1108–1114.
- Słabuszewska-Jóźwiak, A.; Szymański, J.; Ciebiera, M.; Sarecka-Hujar, B.; Jakiel, G. Pediatrics Consequences of Caesarean Section—A Systematic Review and Meta-Analysis. Int. J. Environ. Res. Public Health 2020, 17, 8031.
- Kranich, J.; Maslowski, K.M.; Mackay, C.R. Commensal flora and the regulation of inflammatory and autoimmune responses. Semin. Immunol. 2011, 23, 139–145.
- O’Toole, P.W.; Jeffery, I.B. Gut microbiota and aging. Science 2015, 350, 1214–1215.
- Santoro, A.; Ostan, R.; Candela, M.; Biagi, E.; Brigidi, P.; Capri, M.; Franceschi, C. Gut microbiota changes in the extreme decades of human life: A focus on centenarians. Cell. Mol. Life Sci. 2018, 75, 129–148.
- Biagi, E.; Candela, M.; Franceschi, C.; Brigidi, P. The aging gut microbiota: New perspectives. Ageing Res. Rev. 2011, 10, 428–429.
- Chee, W.J.Y.; Chew, S.Y.; Than, L.T.L. Vaginal microbiota and the potential of Lactobacillus derivatives in maintaining vaginal health. Microb. Cell Factories 2020, 19, 203.
- Kalia, N.; Singh, J.; Kaur, M. Microbiota in vaginal health and pathogenesis of recurrent vulvovaginal infections: A critical review. Ann. Clin. Microbiol. Antimicrob. 2020, 19, 5.
- Tortelli, B.A.; Lewis, W.G.; Allsworth, J.E.; Member-Meneh, N.; Foster, L.R.; Reno, H.E.; Peipert, J.F.; Fay, J.C.; Lewis, A.L. Associations between the vaginal microbiome and Candida colonization in women of reproductive age. Am. J. Obstet. Gynecol. 2020, 222, 471.e1–471.e9.
- Borgogna, J.C.; Shardell, M.D.; Santori, E.K.; Nelson, T.M.; Rath, J.M.; Glover, E.D.; Ravel, J.; Gravitt, P.E.; Yeoman, C.J. The vaginal metabolome and microbiota of cervical HPV-positive and HPV-negative women: A cross-sectional analysis. BJOG Int. J. Obstet. Gynaecol. 2020, 127, 182–192.
- Masha, S.C.; Owuor, C.; Ngoi, J.M.; Cools, P.; Sanders, E.J.; Vaneechoutte, M.; Crucitti, T.; De Villiers, E.P. Comparative analysis of the vaginal microbiome of pregnant women with either Trichomonas vaginalis or Chlamydia trachomatis. PLoS ONE 2019, 14, e0225545.
- Francis, S.C.; Crucitti, T.; Smekens, T.; Hansen, C.H.; Andreasen, A.; Jespers, V.; Hardy, L.; Irani, J.; Changalucha, J.; Baisley, K.; et al. The Vaginal Microbiota Among Adolescent Girls in Tanzania Around the Time of Sexual Debut. Front. Cell. Infect. Microbiol. 2020, 10, 305.
- Barrientos-Durán, A.; Fuentes-López, A.; De Salazar, A.; Plaza-Díaz, J.; García, F. Reviewing the Composition of Vaginal Microbiota: Inclusion of Nutrition and Probiotic Factors in the Maintenance of Eubiosis. Nutrients 2020, 12, 419.
- Greenbaum, S.; Greenbaum, G.; Moran-Gilad, J.; Weintraub, A.Y. Ecological dynamics of the vaginal microbiome in relation to health and disease. Am. J. Obstet. Gynecol. 2019, 220, 324–335.
- Gupta, S.; Kakkar, V.; Bhushan, I. Crosstalk between Vaginal Microbiome and Female Health: A review. Microb. Pathog. 2019, 136, 103696.
- Norenhag, J.; Du, J.; Olovsson, M.; Verstraelen, H.; Engstrand, L.; Brusselaers, N. The vaginal microbiota, human papillomavirus and cervical dysplasia: A systematic review and network meta-analysis. BJOG Int. J. Obstet. Gynaecol. 2020, 127, 171–180.
- Hickey, R.J.; Zhou, X.; Settles, M.L.; Erb, J.; Malone, K.; Hansmann, M.A.; Shew, M.L.; Van Der Pol, B.; Fortenberry, J.D.; Forney, L.J. Vaginal Microbiota of Adolescent Girls Prior to the Onset of Menarche Resemble Those of Reproductive-Age Women. mBio 2015, 6, e00097-15.
- Brown, R.G.; Al-Memar, M.; Marchesi, J.R.; Lee, Y.S.; Smith, A.; Chan, D.; Lewis, H.; Kindinger, L.; Terzidou, V.; Bourne, T.; et al. Establishment of vaginal microbiota composition in early pregnancy and its association with subsequent preterm prelabor rupture of the fetal membranes. Transl. Res. 2019, 207, 30–43.
- Fettweis, J.M.; Serrano, M.G.; Brooks, J.L.; Edwards, D.J.; Girerd, P.H.; Parikh, H.I.; Huang, B.; Arodz, T.J.; Edupuganti, L.; Glascock, A.L.; et al. The vaginal microbiome and preterm birth. Nat. Med. 2019, 25, 1012–1021.
- Hummelen, R.; Macklaim, J.M.; Bisanz, J.E.; Hammond, J.-A.; McMillan, A.; Vongsa, R.; Koenig, D.; Gloor, G.B.; Reid, G. Vaginal Microbiome and Epithelial Gene Array in Post-Menopausal Women with Moderate to Severe Dryness. PLoS ONE 2011, 6, e26602.
- Brotman, R.M.; Shardell, M.D.; Gajer, P.; Fadrosh, D.; Chang, K.; Silver, M.I.; Viscidi, R.P.; Burke, A.E.; Ravel, J.; Gravitt, P.E. Association between the vaginal microbiota, menopause status, and signs of vulvovaginal atrophy. Menopause 2014, 21, 450–458.
- Gliniewicz, K.; Schneider, G.M.; Ridenhour, B.J.; Williams, C.J.; Song, Y.; Farage, M.A.; Miller, K.; Forney, L.J. Comparison of the Vaginal Microbiomes of Premenopausal and Postmenopausal Women. Front. Microbiol. 2019, 10, 193.
- Mitchell, C.M.; Srinivasan, S.; Plantinga, A.; Wu, M.C.; Reed, S.D.; Guthrie, K.A.; LaCroix, A.Z.; Fiedler, T.; Munch, M.; Liu, C.; et al. Associations between improvement in genitourinary symptoms of menopause and changes in the vaginal ecosystem. Menopause 2018, 25, 500–507.
- Sawicki, W.; Malejczyk, J.; Wróblewska, M. Starzenie: Mechanizmy epigenetyczne i genetyczne. Aging: Epigenetic and genetic mechamisms. Ginekol. Pol. 2015, 2, 47–52.
- Truong, T.P.; Sakata-Yanagimoto, M.; Yamada, M.; Nagae, G.; Enami, T.; Nakamoto-Matsubara, R.; Aburatani, H.; Chiba, S. Age-Dependent Decrease of DNA Hydroxymethylation in Human T Cells. J. Clin. Exp. Hematop. 2015, 55, 1–6.
- Li, D.; Guo, B.; Wu, H.; Tan, L.; Lu, Q. TET Family of Dioxygenases: Crucial Roles and Underlying Mechanisms. Cytogenet. Genome Res. 2015, 146, 171–180.
- Li, N.; Chen, J.; Pei, D. The Battle between TET Proteins and DNA Methylation for the Right Cell. Trends Cell Biol. 2018, 28, 973–975.
- Kagiwada, S.; Kurimoto, K.; Hirota, T.; Yamaji, M.; Saitou, M. Replication-coupled passive DNA demethylation for the erasure of genome imprints in mice. EMBO J. 2012, 32, 340–353.
- Parker, M.J.; Weigele, P.R.; Saleh, L. Insights into the Biochemistry, Evolution, and Biotechnological Applications of the Ten-Eleven Translocation (TET) Enzymes. Biochemistry 2019, 58, 450–467.
- Ko, M.; An, J.; Bandukwala, H.S.; Chavez, L.; Äijö, T.; Pastor, W.A.; Segal, M.F.; Li, H.; Koh, K.P.; Lähdesmäki, H.; et al. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature 2013, 497, 122–126.
- Hu, X.; Chen, Y.; Zhao, Z.J. Structure, regulation, and function of TET family proteins. In Epigenetic Gene Expression and Regulation; Huang, S., Litt, M.D., Blakey, C.A., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 379–395.
- Ulrich, C.M.; Toriola, A.T.; Koepl, L.M.; Sandifer, T.; Poole, E.M.; Duggan, C.; McTiernan, A.; Issa, J.-P.J. Metabolic, hormonal and immunological associations with global DNA methylation among postmenopausal women. Epigenetics 2012, 7, 1020–1028.

