 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mohammad Mojtaba Sadeghi | + 3476 word(s) | 3476 | 2021-06-02 11:09:08 | | | |
| 2 | Peter Tang | -14 word(s) | 3462 | 2021-06-09 03:45:08 | | | | |
| 3 | Conner Chen | Meta information modification | 3462 | 2021-09-22 09:43:04 | | |
Video Upload Options
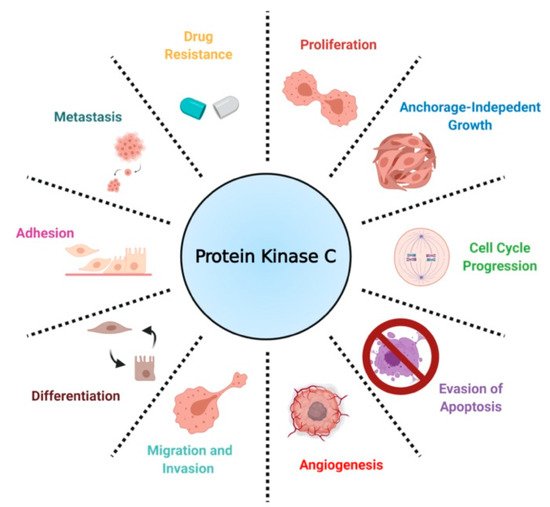
Despite significant advances, targeted therapy is greatly limited by resistance acquisition, which emerges in nearly all patients receiving treatment. As a result, identifying the molecular modulators of resistance is of great interest. Recent work has implicated protein kinase C (PKC) isozymes as mediators of drug resistance in non-small cell lung cancer (NSCLC). Importantly, previous findings on PKC have implicated this family of enzymes in both tumor-promotive and tumor-suppressive biology in various tissues. Here, we review the biological role of PKC isozymes in NSCLC through extensive analysis of cell-line-based studies to better understand the rationale for PKC inhibition.
1. Introduction
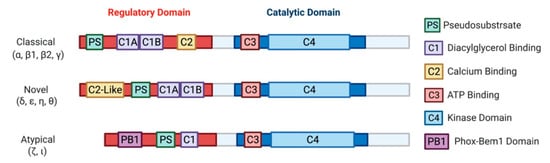
2. The Protein Kinase C Family
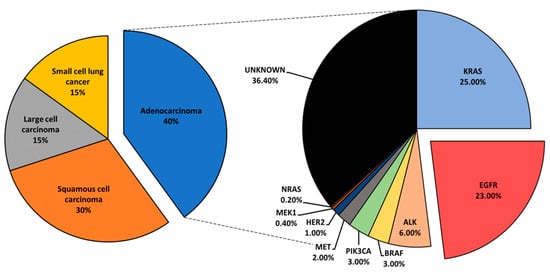
3. Expression, Biological Role, and Prognosis of Protein Kinase C in NSCLC
|
Isozyme |
Biological Roles |
References |
|---|---|---|
|
PKC α |
Promotes proliferation, invasion, migration, cell cycle progression, evasion of apoptosis, drug resistance |
|
|
PKC β1, β2 |
Unknown |
|
|
PKC γ |
Not expressed |
|
|
PKC δ |
Mediates drug sensitivity, invasion, cell survival |
|
|
PKC ε |
Promotes proliferation, invasion, migration, cell cycle progression, anchorage-independent growth, evasion of apoptosis |
|
|
PKC η |
Mediates drug sensitivity |
[44] |
|
PKC θ |
Not expressed |
|
|
PKC ζ |
Chemotaxis |
[71] |
|
PKC ι |
Promotes proliferation, invasion, migration, anchorage-independent growth, evasion of apoptosis, stemness, glucose metabolism |
4. Therapeutic Approaches Targeting PKCs in NSCLC
5. PKC-Mediated Resistance Acquisition and Drug Sensitivity in NSCLC
In addition to the already described biology, PKCs appear to play a role in mediating sensitivity to cytotoxic agents and targeted therapy based on in vitro models. More recent findings suggest that PKCs may also play a crucial role in mediating resistance to tyrosine kinase inhibitors (TKI). One study reported that erlotinib-resistant H1650-M3 cells expressed significantly higher PKCα and had lower PKCδ mRNA levels relative to the parental H1650. RNAi and small molecule inhibition of PKCα sensitized H1650-M3 to erlotinib, a TKI inhibitor of ΔEGFR. Importantly, however, a viral-mediated stable overexpression of PKCα did not affect H1650 sensitivity to erlotinib. This strongly suggests that PKCα alone is not sufficient to induce erlotinib resistance. On the other hand, viral-mediated overexpression of PKCδ moderately increased H1650-M3 sensitivity to erlotinib [82]. A subsequent study combining PKC inhibitor chelerythrine chloride and erlotinib in the treatment of NSCLC A549 and SK-MES1 cells reported a significant synergy in impairing cell viability, colony formation, tumor growth in xenografts, and enhanced apoptosis. Importantly, however, the concentrations of erlotinib used in the experiments (2.5–20 μM) typically exceeded the therapeutic range (1–2 μM), and the cell lines used in the study have wild-type EGFR [83]. A more recent study evaluating EGFR-TKI resistance reported PKCδ to be necessary and sufficient to induce resistance, and downregulation of the kinase via molecular and pharmacological approaches sensitized TKI-resistant cells to erlotinib. Although contradictory to the previous study in H1650-M3, an shRNA-mediated downregulation of PKCδ sensitized H1650 and TKI-resistant HCC827-GR cells to gefitinib, another EGFR TKI. Importantly, ectopic expression of PKCδ was sufficient to induce resistance in TKI-sensitive HCC827 and H3255. Mutations in the nuclear localization signal that sequestered PKCδ in the cytoplasm impaired PKCδ- induced gefitinib resistance and attenuated phosphorylation of ERK, AKT, and RelA in isolated nuclear fractions. Clinically, PKCδ upregulation correlated negatively with OS in TKI-treated ΔEGFR NSCLC patients [84]. Interestingly, our recently published work has demonstrated that there is a ΔEGFR-independent selection for high PKCα protein expression in NSCLC, and a ΔEGFR-dependent activation of PKCα that translates to constitutive downstream signaling to AKT/mTOR pathway [113]. In BRAF/ KRAS mutant NSCLC, PKCα was shown to modulate sensitivity to chemotherapy via mediating MDR1 expression and RLIP76 phosphorylation (Figure 4) [40][41][42]. These findings suggest that PKC α and δ inhibition, in the context of mutant driver genes such as EGFR and KRAS, may be a novel approach to address TKI-sensitivity and resistance to chemotherapy in NSCLC.
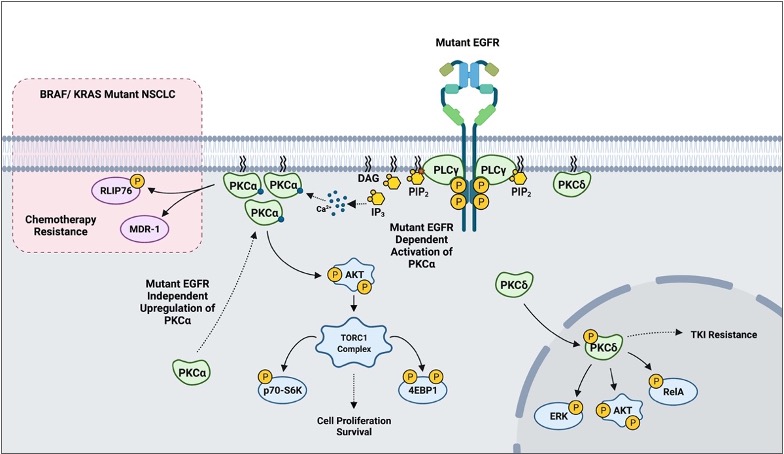
Figure 4. A scheme outlining PKC-mediated drug resistance in EGFR and KRAS mutant NSCLC.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34.
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594.
- Zheng, M. Classification and pathology of lung cancer. Surg. Oncol. Clin. 2016, 25, 447–468.
- Cruz, C.S.; Tanoue, L.T.; Matthay, R.A. Lung cancer: Epidemiology, etiology, and prevention. Clin. Chest Med. 2011, 32, 605–644.
- Kris, M.G.; Johnson, B.E.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Aronson, S.L.; Engelman, J.A.; Shyr, Y.; Khuri, F.R.; Rudin, C.M.; et al. Identification of driver mutations in tumor specimens from 1000 patients with lung adenocarcinoma: The NCI’s Lung Cancer Mutation Consortium (LCMC). J. Clin. Oncol. 2011, 29, CRA7506.
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as firstline treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer: A multicentre, open-label, randomized, phase 3 study. Lancet Oncol. 2011, 12, 735–742.
- Jackman, D.; Pao, W.; Riely, G.J.; Engelman, J.A.; Kris, M.G.; Jänne, P.A.; Lynch, T.; Johnson, B.E.; Miller, V.A. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 357–360.
- Kang, J.H. Protein kinase C (PKC) isozymes and cancer. New J. Sci. 2014, 2014, 1–36.
- Singh, R.M.; Cummings, E.; Pantos, C.; Singh, J. Protein kinase C and cardiac dysfunction: A review. Heart Fail. Rev. 2017, 22, 843–859.
- Marrocco, V.; Bogomolovas, J.; Ehler, E.; dos Remedios, C.G.; Yu, J.; Gao, C.; Lange, S. PKC and PKN in heart disease. J. Mol. Cell. Cardiol. 2019, 128, 212–226.
- Evcimen, N.D.; King, G.L. The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol. Res. 2007, 55, 498–510.
- Geraldes, P.; King, G.L. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ. Res. 2010, 106, 1319–1331.
- Saxena, A.; Scaini, G.; Bavaresco, D.V.; Leite, C.; Valvassoria, S.S.; Carvalho, A.F.; Quevedo, J. Role of protein kinase C in bipolar disorder: A review of the current literature. Mol. Neuropsychiatry 2017, 3, 108–124.
- Alfonso, S.I.; Callender, J.A.; Hooli, B.; Antal, C.E.; Mullin, K.; Sherman, M.A.; Lesné, S.E.; Leitges, M.; Newton, A.; Tanzi, R.E.; et al. Gain-of function mutations in protein kinase Cα (PKCα) may promote synaptic defects in Alzheimer’s disease. Sci. Signal. 2016, 9, ra47.
- Koivunen, J.; Aaltonen, V.; Peltonen, J. Protein kinase C (PKC) family in cancer progression. Cancer Lett. 2006, 235, 1–10.
- Newton, A.C. Protein kinase C as a tumor suppressor. Semin. Cancer Biol. 2018, 48, 18–26.
- Inoue, M.; Kishimoto, A.; Takai, Y.; Nishizuka, Y. Studies on a cyclic nucleotide-independent protein kinase and its proenzyme in mammalian tissues. II. Proenzyme and its activation by calcium-dependent protease from rat brain. J. Biol. Chem. 1977, 252, 7610–7616.
- Parker, P.J.; Coussens, L.; Totty, N.; Rhee, L.; Young, S.; Chen, E.; Stabel, S.; Waterfield, M.D.; Ullrich, A. The complete primary structure of protein kinase C—The major phorbol ester receptor. Science 1986, 233, 853–859.
- Coussens, L.; Parker, P.J.; Rhee, L.; Yang-Feng, T.L.; Chen, E.; Waterfield, M.D.; Francke, U.; Ullrich, A. Multiple, distinct forms of bovine and human protein kinase C suggest diversity in cellular signaling pathways. Science 1986, 233, 859–866.
- Jaken, S.; Kiley, S.C. Purification and characterization of three types of protein kinase C from rabbit brain cytosol. Proc. Natl. Acad. Sci. USA 1987, 84, 4418–4422.
- Ohno, S.; Kawasaki, H.; Imajoh, S.; Suzuki, K.; Inagaki, M.; Yokokura, H.; Sakoh, T.; Hidaka, H. Tissue-specific expression of three distinct types of rabbit protein kinase C. Nature 1987, 325, 161.
- Kubo, K.; Ohno, S.; Suzuki, K. Primary structures of human protein kinase C βI and βII differ only in their C-terminal sequences. FEBS Lett. 1987, 223, 138–142.
- Ono, Y.; Fuji, T.; Ogita, K.; Kikkawa, U.; Igarashi, K.; Nishizuka, Y. Identification of three additional members of rat protein kinase C family: δ-, ϵ- and ξ-subspecies. FEBS Lett. 1987, 226, 125–128.
- Osada, S.I.; Mizuno, K.; Saido, T.C.; Akita, Y.; Suzuki, K.; Kuroki, T.; Ohno, S. A phorbol ester receptor/protein kinase, nPKC eta, a new member of the protein kinase C family predominantly expressed in lung and skin. J. Biol. Chem. 1990, 265, 22434–22440.
- Steinberg, S.F. Structural basis of protein kinase C isoform function. Physiol. Rev. 2008, 88, 1341–1378.
- Takai, Y.; Kishimoto, A.; Kikkawa, U.; Mori, T.; Nishizuka, Y. Unsaturated diacylglycerol as a possible messenger for the activation of calcium-activated, phospholipid-dependent protein kinase system. Biochem. Biophys. Res. Commun. 1979, 91, 1218–1224.
- Castagna, M.; Takai, Y.; Kaibuchi, K.; Sano, K.; Kikkawa, U.; Nishizuka, Y. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J. Biol. Chem. 1982, 257, 7847–7851.
- Antal, C.E.; Hudson, A.M.; Kang, E.; Zanca, C.; Wirth, C.; Stephenson, N.L.; Trotter, E.W.; Gallegos, L.L.; Miller, C.J.; Furnari, F.B.; et al. Cancer-associated protein kinase C mutations reveal kinase’s role as tumor suppressor. Cell 2015, 160, 489–502.
- Newton, A.C. Protein kinase C: Perfectly balanced. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 208–230.
- Isakov, N. Protein kinase C (PKC) isoforms in cancer, tumor promotion and tumor suppression. Semin. Cancer Biol. 2018, 48, 36–52.
- Lahn, M.; Su, C.; Li, S.; Chedid, M.; Hanna, K.R.; Graff, J.R.; Sandusky, G.E.; Ma, D.; Niyikiza, C.; Sundell, K.L.; et al. Expression levels of protein kinase C-α in non-small-cell lung cancer. Clin. Lung Cancer 2004, 6, 184–189.
- Clark, A.S.; West, K.A.; Blumberg, P.M.; Dennis, P.A. Altered protein kinase C (PKC) isoforms in non-small cell lung cancer cells: PKCδ promotes cellular survival and chemotherapeutic resistance. Cancer Res. 2003, 63, 780–786.
- Hirai, M.; Gamou, S.; Kobayashi, M.; Shimizu, N. Lung cancer cells often express high levels of protein kinase C activity. Jpn. J. Cancer Res. 1989, 80, 204–208.
- Jiang, H.; Fu, Q.; Song, X.; Ge, C.; Li, R.; Li, Z.; Zeng, B.; Li, C.; Wang, Y.; Xue, Y.; et al. HDGF and PRKCA upregulation is associated with a poor prognosis in patients with lung adenocarcinoma. Oncol. Lett. 2019, 18, 4936–4946.
- Wang, X.Y.; Repasky, E.; Liu, H.T. Antisense inhibition of protein kinase Cα reverses the transformed phenotype in human lung carcinoma cells. Exp. Cell Res. 1999, 250, 253–263.
- Wang, C.; Wang, X.; Liang, H.; Wang, T.; Yan, X.; Cao, M.; Wang, N.; Zhang, S.; Zen, K.; Zhang, C.; et al. miR-203 inhibits cell proliferation and migration of lung cancer cells by targeting PKCα. PLoS ONE 2013, 8, e73985.
- Dean, N.; McKay, R.; Miraglia, L.; Howard, R.; Cooper, S.; Giddings, J.; Nicklin, P.; Meister, L.; Ziel, R.; Geiger, T.; et al. Inhibition of growth of human tumor cell lines in nude mice by an antisense oligonucleotide inhibitor of protein kinase C-α expression. Cancer Res. 1996, 56, 3499–3507.
- Deeds, L.; Teodorescu, S.; Chu, M.; Yu, Q.; Chen, C.Y. A p53-independent G1 cell cycle checkpoint induced by the suppression of protein kinase C α and θ isoforms. J. Biol. Chem. 2003, 278, 39782–39793.
- Singhal, S.S.; Wickramarachchi, D.; Singhal, J.; Yadav, S.; Awasthi, Y.C.; Awasthi, S. Determinants of differential doxorubicin sensitivity between SCLC and NSCLC. FEBS Lett. 2006, 580, 2258–2264.
- Singhal, S.S.; Yadav, S.; Singhal, J.; Awasthi, Y.C.; Awasthi, S. Mitogenic and drug-resistance mediating effects of PKCα require RLIP76. Biochem. Biophys. Res. Commun. 2006, 348, 722–727.
- Wang, X.Y.; Liu, H.T. Antisense expression of protein kinase C alpha improved sensitivity to anticancer drugs in human lung cancer LTEPa-2 cells. Acta Pharmacol. Sin. 1998, 19, 265–268.
- Gao, Z.; Tang, L.; Su, B.; Sha, H.; Han, B. Effects of protein kinase C inhibitor, chelerythrine chloride, on drug-sensitivity of NSCLC cell lines. Chin. J. Lung Cancer 2007, 1, 455–460.
- Sonnemann, J.; Gekeler, V.; Ahlbrecht, K.; Brischwein, K.; Liu, C.; Bader, P.; Müller, C.; Niethammer, D.; Beck, J.F. Down-regulation of protein kinase Cη by antisense oligonucleotides sensitizes A549 lung cancer cells to vincristine and paclitaxel. Cancer Lett. 2004, 209, 177–185.
- Kang, J.H.; Mori, T.; Kitazaki, H.; Niidome, T.; Takayama, K.; Nakanishi, Y.; Katayama, Y. Kinase activity of protein kinase cα in serum as a diagnostic biomarker of human lung cancer. Anticancer Res. 2013, 33, 485–488.
- Symonds, J.M.; Ohm, A.M.; Carter, C.J.; Heasley, L.E.; Boyle, T.A.; Franklin, W.A.; Reyland, M.E. Protein kinase C δ is a downstream effector of oncogenic K-ras in lung tumors. Cancer Res. 2011, 71, 2087–2097.
- Kim, E.H.; Lee, H.J.; Lee, D.H.; Bae, S.; Soh, J.W.; Jeoung, D.; Kim, J.; Cho, C.K.; Lee, Y.J.; Lee, Y.S. Inhibition of heat shock protein 27-mediated resistance to DNA damaging agents by a novel PKC delta-V5 heptapeptide. Cancer Res. 2007, 67, 6333–6341.
- Bae, K.M.; Wang, H.; Jiang, G.; Chen, M.G.; Lu, L.; Xiao, L. Protein Kinase Cε Is Overexpressed in Primary Human Non-Small Cell Lung Cancers and Functionally Required for Proliferation of Non-Small Cell Lung Cancer Cells in a p21/Cip1-Dependent Manner. Cancer Res. 2007, 67, 6053–6063.
- Totoń, E.; Ignatowicz, E.; Skrzeczkowska, K.; Rybczyńska, M. Protein kinase Cε as a cancer marker and target for anticancer therapy. Pharmacol. Rep. 2011, 63, 19–29.
- Caino, M.C.; Lopez-Haber, C.; Kim, J.; Mochly-Rosen, D.; Kazanietz, M.G. Protein kinase Cɛ is required for non-small cell lung carcinoma growth and regulates the expression of apoptotic genes. Oncogene 2012, 31, 2593–2600.
- Caino, M.C.; Lopez-Haber, C.; Kissil, J.L.; Kazanietz, M.G. Non-small cell lung carcinoma cell motility, rac activation and metastatic dissemination are mediated by protein kinase C epsilon. PLoS ONE 2012, 7, e31714.
- Zhang, N.; Su, Y.; Xu, L. Targeting PKCε by miR-143 regulates cell apoptosis in lung cancer. FEBS Lett. 2013, 587, 3661–3667.
- Zurgil, U.; Ben-Ari, A.; Rotem-Dai, N.; Karp, G.; Krasnitsky, E.; Frost, S.A.; Livneh, E. PKCη is an anti-apoptotic kinase that predicts poor prognosis in breast and lung cancer. Biochem. Soc. Trans. 2014, 42, 1519–1523.
- Krasnitsky, E.; Baumfeld, Y.; Freedman, J.; Sion-Vardy, N.; Ariad, S.; Novack, V.; Livneh, E. PKCη is a novel prognostic marker in non-small cell lung cancer. Anticancer Res. 2012, 32, 1507–1513.
- Basu, A. The Enigmatic Protein Kinase C-eta. Cancers 2019, 11, 214.
- Luo, Q.; Tang, L.; Lin, H.; Huang, J.; Zhang, T.; Liu, Y.; Wang, J.; Zhan, P.; Yin, X.; Su, X.; et al. The oncogenic role of PKCiota gene amplification and overexpression in Chinese non-small cell lung cancer. Lung Cancer 2014, 84, 190–195.
- Regala, R.P.; Weems, C.; Jamieson, L.; Khoor, A.; Edell, E.S.; Lohse, C.M.; Fields, A.P. Atypical protein kinase Cι is an oncogene in human non-small cell lung cancer. Cancer Res. 2005, 65, 8905–8911.
- Fields, A.P.; Regala, R.P. Protein kinase Cι: Human oncogene, prognostic marker and therapeutic target. Pharmacol. Res. 2007, 55, 487–497.
- BommaReddy, R.R.; Patel, R.; Smalley, T.; Acevedo-Duncan, M. Effects of atypical protein kinase C inhibitor (DNDA) on lung cancer proliferation and migration by PKC-ι/FAK ubiquitination through the Cbl-b pathway. OncoTargets Ther. 2020, 13, 1661.
- Kim, K.H.; Chung, C.; Kim, J.M.; Lee, D.; Cho, S.Y.; Lee, T.H.; Cho, H.J.; Yeo, M.K. Clinical significance of atypical protein kinase C (PKCι and PKCζ) and its relationship with yes-associated protein in lung adenocarcinoma. BMC Cancer 2019, 19, 804.
- Regala, R.P.; Weems, C.; Jamieson, L.; Copland, J.A.; Thompson, E.A.; Fields, A.P. Atypical protein kinase Cι plays a critical role in human lung cancer cell growth and tumorigenicity. J. Biol. Chem. 2005, 280, 31109–31115.
- Imamura, N.; Horikoshi, Y.; Matsuzaki, T.; Toriumi, K.; Kitatani, K.; Ogura, G.; Masuda, R.; Nakamura, N.; Takekoshi, S.; Iwazaki, M. Localization of aPKC lambda/iota and its interacting protein, Lgl2, is significantly associated with lung adenocarcinoma progression. Tokai J. Exp. Clin. Med. 2013, 38, 146–158.
- Liu, L.; Lei, B.; Wang, L.; Chang, C.; Yang, H.; Liu, J.; Huang, G.; Xie, W. Protein kinase C-iota-mediated glycolysis promotes non-small-cell lung cancer progression. OncoTargets Ther. 2019, 12, 5835–5848.
- Erdogan, E.; Klee, E.W.; Thompson, E.A.; Fields, A.P. Meta-analysis of oncogenic protein kinase Cι signaling in lung adenocarcinoma. Clin. Cancer Res. 2009, 15, 1527–1533.
- Justilien, V.; Jameison, L.; Der, C.J.; Rossman, K.L.; Fields, A.P. Oncogenic activity of Ect2 is regulated through protein kinase Cι-mediated phosphorylation. J. Biol. Chem. 2011, 286, 8149–8157.
- Shultz, J.C.; Vu, N.; Shultz, M.D.; Mba, M.U.; Shapiro, B.A.; Chalfant, C.E. The Proto-oncogene PKCι regulates the alternative splicing of Bcl-x pre-mRNA. Mol. Cancer Res. 2012, 10, 660–669.
- Ali, S.A.; Justilien, V.; Jamieson, L.; Murray, N.R.; Fields, A.P. Protein kinase Cι drives a NOTCH3-dependent stem-like phenotype in mutant KRAS lung adenocarcinoma. Cancer Cell 2016, 29, 367–378.
- Fields, A.P.; Ali, S.A.; Justilien, V.; Murray, N.R. Targeting oncogenic protein kinase Cι for treatment of mutant KRAS LADC. Small GTPases 2017, 8, 58–64.
- Ito, M.; Codony-Servat, C.; Codony-Servat, J.; Lligé, D.; Chaib, I.; Sun, X.; Miao, J.; Sun, R.; Cai, X.; Verlicchi, A.; et al. Targeting PKCι-PAK1 signaling pathways in EGFR and KRAS mutant adenocarcinoma and lung squamous cell carcinoma. Cell Commun. Signal. 2019, 17, 137.
- Yin, N.; Liu, Y.; Khoor, A.; Wang, X.; Thompson, E.A.; Leitges, M.; Justilien, V.; Weems, C.; Murray, N.R.; Fields, A.P. Protein Kinase Cι and Wnt/β-Catenin Signaling: Alternative Pathways to Kras/Trp53-Driven Lung Adenocarcinoma. Cancer Cell 2019, 36, 156–167.
- Liu, Y.; Wang, B.; Wang, J.; Wan, W.; Sun, R.; Zhao, Y.; Zhang, N. Down-regulation of PKCζ expression inhibits chemotaxis signal transduction in human lung cancer cells. Lung Cancer 2009, 63, 210–218.
- Teicher, B.A.; Menon, K.; Alvarez, E.; Shih, C.; Faul, M.M. Antiangiogenic and antitumor effects of a protein kinase Cβ inhibitor in human breast cancer and ovarian cancer xenografts. Investig. New Drugs 2002, 20, 241–251.
- Carmo, A.D.; Balça-Silva, J.; Matias, D.; Lopes, M. PKC signaling in glioblastoma. Cancer Biol. Ther. 2013, 14, 287–294.
- Kim, J.; Choi, Y.L.; Vallentin, A.; Hunrichs, B.S.; Hellerstein, M.K.; Peehl, D.M.; Mochly-Rosen, D. Centrosomal PKCβII and pericentrin are critical for human prostate cancer growth and angiogenesis. Cancer Res. 2008, 68, 6831–6839.
- Kazi, J.U.; Kabir, N.N.; Rönnstrand, L. Protein kinase C (PKC) as a drug target in chronic lymphocytic leukemia. Med. Oncol. 2013, 30, 757.
- Zum Bueschenfelde, C.M.; Wagner, M.; Lutzny, G.; Oelsner, M.; Feuerstacke, Y.; Decker, T.; Bogner, C.; Peschel, C.; Ringshausen, I. Recruitment of PKC-βII to lipid rafts mediates apoptosis-resistance in chronic lymphocytic leukemia expressing ZAP-70. Leukemia 2010, 24, 141–152.
- Murray, N.R.; Baumgardner, G.P.; Burns, D.J.; Fields, A.P. Protein kinase C isotypes in human erythroleukemia (K562) cell proliferation and differentiation. Evidence that beta II protein kinase C is required for proliferation. J. Biol. Chem. 1993, 268, 15847–15853.
- Lutzny, G.; Kocher, T.; Schmidt-Supprian, M.; Rudelius, M.; Klein-Hitpass, L.; Finch, A.J.; Dürig, J.; Wagner, M.; Haferlach, C.; Kohlmann, A.; et al. Protein kinase c-β-dependent activation of NF-κB in stromal cells is indispensable for the survival of chronic lymphocytic leukemia B cells in vivo. Cancer Cell 2013, 23, 77–92.
- Wetsel, W.C.; Khan, W.A.; Merchenthaler, I.; Rivera, H.; Halpern, A.E.; Phung, H.M.; Negro-Vilar, A.; Hannun, Y.A. Tissue and cellular distribution of the extended family of protein kinase C isoenzymes. J. Cell. Biol. 1992, 117, 121–133.
- Brezar, V.; Tu, W.J.; Seddiki, N. PKC-theta in regulatory and effector T-cell functions. Front. Immunol. 2015, 6, 530.
- Baier, G.; Telford, D.; Giampa, L.; Coggeshall, K.M.; Baier-Bitterlich, G.; Isakov, N.; Altman, A. Molecular cloning and characterization of PKC theta, a novel member of the protein kinase C (PKC) gene family expressed predominantly in hematopoietic cells. J. Biol. Chem. 1993, 268, 4997–5004.
- Abera, M.B.; Kazanietz, M.G. Protein kinase Cα mediates erlotinib resistance in lung cancer cells. Mol. Pharmacol. 2015, 87, 832–841.
- He, M.; Yang, Z.; Le Zhang, C.S.; Li, Y.; Zhang, X. Additive effects of cherlerythrine chloride combination with erlotinib in human non-small cell lung cancer cells. PLoS ONE 2017, 12, e0175466.
- Lee, P.C.; Fang, Y.F.; Yamaguchi, H.; Wang, W.J.; Chen, T.C.; Hong, X.; Ke, B.; Xia, W.; Wei, Y.; Zha, Z.; et al. Targeting PKCδ as a therapeutic strategy against heterogeneous mechanisms of EGFR inhibitor resistance in EGFR-mutant lung cancer. Cancer Cell 2018, 34, 954–969.
- Graff, J.R.; McNulty, A.M.; Hanna, K.R.; Konicek, B.W.; Lynch, R.L.; Bailey, S.N.; Banks, C.; Capen, A.; Goode, R.; Lewis, J.E.; et al. The protein kinase Cβ–selective inhibitor, enzastaurin (LY317615. HCl), suppresses signaling through the AKT pathway, induces apoptosis, and suppresses growth of human colon cancer and glioblastoma xenografts. Cancer Res. 2005, 65, 7462–7469.
- Hanauske, A.R.; Oberschmidt, O.; Hanauske-Abel, H.; Lahn, M.M.; Eismann, U. Antitumor activity of enzastaurin (LY317615. HCl) against human cancer cell lines and freshly explanted tumors investigated in vitro soft-agar cloning experiments. Investig. New Drugs 2007, 25, 205–210.
- Mukohara, T.; Nagai, S.; Koshiji, M.; Yoshizawa, K.; Minami, H. Phase I dose escalation and pharmacokinetic study of oral enzastaurin (LY317615) in advanced solid tumors. Cancer Sci. 2010, 101, 2193–2199.
- Carducci, M.A.; Musib, L.; Kies, M.S.; Pili, R.; Truong, M.; Brahmer, J.R.; Cole, P.; Sullivan, R.; Riddle, J.; Schimidt, J.; et al. Phase I dose escalation and pharmacokinetic study of enzastaurin, an oral protein kinase C beta inhibitor, in patients with advanced cancer. J. Clin. Oncol. 2006, 24, 4092–4099.
- Oh, Y.; Herbst, R.S.; Burris, H.; Cleverly, A.; Musib, L.; Lahn, M.; Bepler, G. Enzastaurin, an Oral Serine/Threonine Kinase Inhibitor, As Second- or Third-Line Therapy of Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2008, 26, 1135–1141.
- Tekle, C.; Giovannetti, E.; Sigmond, J.; Graff, J.R.; Smid, K.; Peters, G.J. Molecular pathways involved in the synergistic interaction of the PKCβ inhibitor enzastaurin with the antifolate pemetrexed in non-small cell lung cancer cells. Br. J. Cancer 2008, 99, 750–759.
- Tanai, C.; Yamamoto, N.; Ohe, Y.; Takahashi, T.; Kunitoh, H.; Murakami, H.; Yamamoto, N.; Nakamura, Y.; Nokihara, H.; Shukuya, T.; et al. A phase I study of enzastaurin combined with pemetrexed in advanced non-small cell lung cancer. J. Thorac. Oncol. 2010, 5, 1068–1074.
- Chiappori, A.; Bepler, G.; Barlesi, F.; Soria, J.C.; Reck, M.; Bearz, A.; Barata, F.; Scagliotti, G.; Park, K.; Wagle, A.; et al. Phase II, double-blinded, randomized study of enzastaurin plus pemetrexed as second-line therapy in patients with advanced non-small cell lung cancer. J. Thorac. Oncol. 2010, 5, 369–375.
- Vansteenkiste, J.; Ramlau, R.; Von Pawel, J.; San Antonio, B.; Eschbach, C.; Szczesna, A.; Kennedy, L.; Visseren-Grul, C.; Chouaki, N.; Reck, M. A phase II randomized study of cisplatin-pemetrexed plus either enzastaurin or placebo in chemonaive patients with advanced non-small cell lung cancer. Oncology 2012, 82, 25–29.
- Socinski, M.A.; Raju, R.N.; Stinchcombe, T.; Kocs, D.M.; Couch, L.S.; Barrera, D.; Rousey, S.R.; Choksi, J.K.; Jotte, R.; Patt, D.A.; et al. Randomized, phase II trial of pemetrexed and carboplatin with or without enzastaurin versus docetaxel and carboplatin as first-line treatment of patients with stage IIIB/IV non-small cell lung cancer. J. Thorac. Oncol. 2010, 5, 1963–1969.
- Zhang, L.L.; Cao, F.F.; Wang, Y.; Meng, F.L.; Zhang, Y.; Zhong, D.S.; Zhou, Q.H. The protein kinase C (PKC) inhibitors combined with chemotherapy in the treatment of advanced non-small cell lung cancer: Meta-analysis of randomized controlled trials. Clin. Transl. Oncol. 2015, 17, 371–377.
- Newton, A.C.; Brognard, J. Reversing the Paradigm: Protein Kinase C as a Tumor Suppressor. Trends Pharmacol. Sci. 2017, 38, 438–447.
- Lu, Z.; Liu, D.; Hornia, A.; Devonish, W.; Pagano, M.; Foster, D.A. Activation of protein kinase C triggers its ubiquitination and degradation. Mol. Cell. Biol. 1998, 18, 839–845.
- Young, S.; Parker, P.J.; Ullrich, A.; Stabel, S. Down-regulation of protein kinase C is due to an increased rate of degradation. Biochem. J. 1987, 244, 775–779.
- Gescher, A.; Reed, D.J. Characterization of the growth inhibition induced by tumor-promoting phorbol esters and of their receptor binding in A549 human lung carcinoma cells. Cancer Res. 1985, 45, 4315–4321.
- Dale, I.L.; Gescher, A. Effects of activators of protein kinase C, including bryostatins 1 and 2, on the growth of A549 human lung carcinoma cells. Int. J. Cancer 1989, 43, 158–163.
- Dale, I.L.; Bradshaw, T.D.; Gescher, A.; Pettit, G.R. Comparison of effects of bryostatins 1 and 2 and 12-O-tetradecanoylphorbol-13-acetate on protein kinase C activity in A549 human lung carcinoma cells. Cancer Res. 1989, 49, 3242–3245.
- Bradshaw, T.D.; Gescher, A.; Pettit, G.R. Modulation by staurosporine of phorbol-ester-induced effects on growth and protein kinase C localization in A549 human lung-carcinoma cells. Int. J. Cancer 1992, 51, 144–148.
- Tanwell, C.S.; Gescher, A.; Bradshaw, T.D.; Pettit, G.R. The role of protein kinase C isoenzymes in the growth inhibition caused by bryostatin 1 in human A549 lung and MCF-7 breast carcinoma cells. Int. J. Cancer 1994, 56, 585–592.
- Tahara, E.; Kadara, H.; Lacroix, L.; Lotan, D.; Lotan, R. Activation of protein kinase C by phorbol 12-myristate 13-acetate suppresses the growth of lung cancer cells through KLF6 induction. Cancer Biol. Ther. 2009, 8, 801–807.
- Xiao, L.; Caino, M.C.; von Burstin, V.A.; Oliva, J.L.; Kazanietz, M.G. Phorbol Ester-Induced Apoptosis and Senescence in Cancer Cell Models. Methods Enzymol. 2008, 446, 123–139.
- Hornung, R.L.; Pearson, J.W.; Beckwith, M.; Longo, D.L. Preclinical evaluation of bryostatin as an anticancer agent against several murine tumor cell lines: In vitro versus in vivo activity. Cancer Res. 1992, 52, 101–107.
- Winegarden, J.D.; Mauer, A.M.; Gajewski, T.F.; Hoffman, P.C.; Krauss, S.; Rudin, C.M.; Vokes, E.E. A phase II study of bryostatin-1 and paclitaxel in patients with advanced non-small cell lung cancer. Lung Cancer 2003, 39, 191–196.
- Geiger, T.; Müller, M.; Dean, N.M.; Fabbro, D. Antitumor activity of a PKC-alpha antisense oligonucleotide in combination with standard chemotherapeutic agents against various human tumors transplanted into nude mice. Anticancer Drug Des. 1998, 13, 35–45.
- Villalona-Calero, M.A.; Ritch, P.; Figueroa, J.A.; Otterson, G.A.; Belt, R.; Dow, E.; George, S.; Leonardo, J.; McCachren, S.; Miller, G.L.; et al. A Phase I/II study of LY900003, an antisense inhibitor of protein kinase C-α, in combination with cisplatin and gemcitabine in patients with advanced non-small cell lung cancer. Clin. Cancer Res. 2004, 10, 6086–6093.
- Ritch, P.; Rudin, C.M.; Bitran, J.D.; Edelman, M.J.; Makalinao, A.; Irwin, D.; Lilenbaum, R.; Peterson, P.; John, W.J. Phase II study of PKC-α antisense oligonucleotide aprinocarsen in combination with gemcitabine and carboplatin in patients with advanced non-small cell lung cancer. Lung Cancer 2006, 52, 173–180.
- Lynch, T.J.; Raju, R.; Lind, M.; Riviere, A.; Gatzemeier, U.; Dorr, A.; Holmund, J.; Yuen, A.; Sikic, B. Randomized phase III trial of chemotherapy and antisense oligonucleotide LY900003 (ISIS 3521) in patients with advanced NSCLC. Lung Cancer 2003, 41, S35.
- Paz-Ares, L.; Douillard, J.Y.; Koralewski, P.; Manegold, C.; Smit, E.F.; Reyes, J.M.; Chang, G.; John, W.J.; Peterson, P.M.; Obasaju, C.K.; et al. Phase III study of gemcitabine and cisplatin with or without aprinocarsen, a protein kinase C-alpha antisense oligonucleotide, in patients with advanced-stage non-small-cell lung cancer. J. Clin. Oncol. 2006, 24, 1428–1434.
- Salama, M.F.; Liu, M.; Clarke, C.J.; Espaillat, M.P.; Haley, J.D.; Jin, T.; Wang, D.; Obeid, L.M.; Hannun, Y.A. PKCα is required for Akt-mTORC1 activation in non-small cell lung carcinoma (NSCLC) with EGFR mutation. Oncogene 2019, 38, 7311–7328.

