 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ryota Tamura | + 4466 word(s) | 4466 | 2021-06-03 12:36:47 | | | |
| 2 | Ron Wang | Meta information modification | 4466 | 2021-06-08 09:26:27 | | |
Video Upload Options
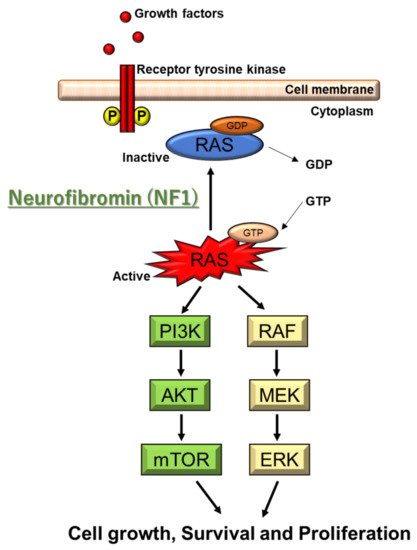
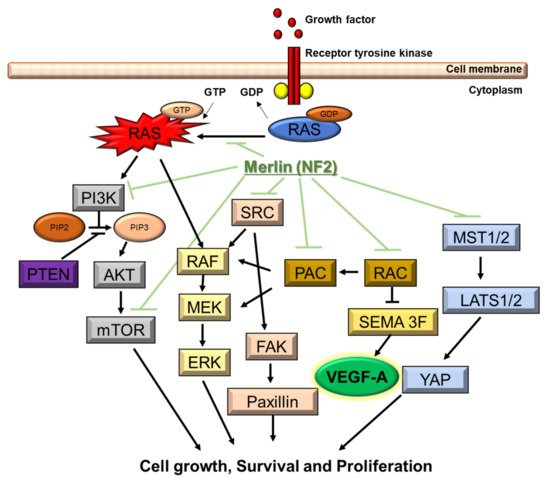
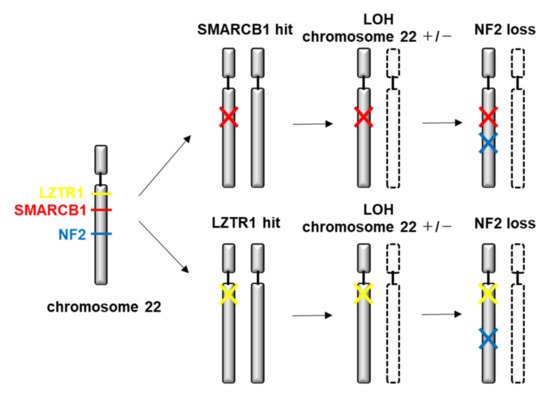
Neurofibromatosis (NF) is a neurocutaneous syndrome characterized by the development of tumors of the central or peripheral nervous system including the brain, spinal cord, organs, skin, and bones. There are three types of NF: NF1 accounting for 96% of all cases, NF2 in 3%, and schwannomatosis (SWN) in <1%. The NF1 gene is located on chromosome 17q11.2, which encodes for a tumor suppressor protein, neurofibromin, that functions as a negative regulator of Ras/MAPK and PI3K/mTOR signaling pathways. The NF2 gene is identified on chromosome 22q12, which encodes for merlin, a tumor suppressor protein related to ezrin-radixin-moesin that modulates the activity of PI3K/AKT, Raf/MEK/ERK, and mTOR signaling pathways. In contrast, molecular insights on the different forms of SWN remain unclear. Inactivating mutations in the tumor suppressor genes SMARCB1 and LZTR1 are considered responsible for a majority of cases.
1. Introduction
2. Neurofibromatosis Type 1
2.1. Clinical Characteristics
| A: The Diagnostic Criteria for NF1 Are Met in an Individual Who Does Not Have a Parent Diagnosed with NF1 if Two or More of the Following Are Present: |
| At least six café-au-lait macules (>5 mm diameter in prepubertal individuals and >15 mm in postpubertal individuals) |
| Freckling in axillary or inguinal regions #1 |
| Optic glioma |
| At least two Lisch nodules identified by slit lamp examination or two or more choroidal abnormalities—defined as bright, patchy nodules imaged by optical coherence tomography/near-infrared reflectance imaging |
| At least two neurofibromas of any type, or one plexiform neurofibroma |
| A distinctive osseous lesion such as sphenoid dysplasia, #2 anterolateral bowing of the tibia, or pseudarthrosis of a long bone |
| A heterozygous pathogenic NF1 variant with a variant allele fraction of 50% in apparently normal tissue such as white blood cells |
| B: A child of a parent who meets the diagnostic criteria specified in A merits a diagnosis of NF1 if one or more of the criteria in A are present |
2.2. Genetic and Molecular Characteristics
2.3. Therapeutic Strategies
2.4. Ongoing Clinical Trials
| ID | Initiation Date | Phase | Nation | N | Disease | Treatment | Primary Outcome |
|---|---|---|---|---|---|---|---|
| NCT04495127 | 8, 2020 | 1 | Japan | 12 | NF1 | Selumetinib | Toxicity |
| NCT01968590 | 8, 2017 | 2 | USA | 320 | NF1 | Cholecalciferol | Bone mineral density |
| NCT03962543 | 9, 2019 | 2 | USA | 100 | NF1 Plexiform Neurofibroma |
Mirdametinib (PD-0325901) oral capsule | Complete or partial response rate compared to baseline. |
| NCT03231306 | 11, 2017 | 2 | USA | 40 | NF1 Plexiform Neurofibroma |
Binimetinib | Change from Baseline Target Tumor Volume at 12 months |
| NCT02839720 | 4, 2017 | 2 | USA | 24 | Cutaneous Neurofibroma NF1 Optic Nerve Glioma |
Selumetinib | Change in the size |
| NCT02407405 | 1, 2016 | 2 | USA | 60 | NF1 Plexiform Neurofibromas |
Selumetinib | Determine objective response rate |
| NCT04461886 | 7, 2020 | 3 | Japan | 100 | NF | NPC-12G gel | Discontinuation rate associated with adverse events |
| NCT03871257 | 10, 2019 | 3 | USA | 290 | Low Grade Glioma NF1 Visual Pathway Glioma |
Carboplatin Selumetinib Sulfate Vincristine Sulfate |
Event-free survival |
| NCT02101736 | 6, 2014 | 2 | USA | 48 | NF1 Neurofibromatosis Plexiform Neurofibromas |
Cabozantinib | The change in tumor size based on radiographic assessment |
| NCT03326388 | 9, 2019 | 1/2 | USA | 30 | NF1 Plexiform Neurofibroma Optic Nerve Glioma |
Selumetinib | To evaluate the Maximum Tolerated Dose Objective response rate |
| NCT03741101 | 6, 2019 | 2 | Sweden | 15 | NF1 Plexiform Neurofibromas |
Trametinib | Remission of tumor volume ≥20% |
| NCT02728388 | 8, 2016 | 2 | USA | 30 | NF1 | aminolevulinic acid | Time to disease progression |
| NCT04435665 | 8, 2020 | 2 | USA | 48 | NF1 Cutaneous Neurofibroma |
NFX-179 Gel | Phospho-erk (p-ERK) levels of Target cNF Tumors Toxicity |
| NCT02390752 | 4, 2015 | 1/2 | USA | 81 | Neurofibroma, Plexiform | PLX3397 | Toxicity Objective response rate |
| NCT03688568 | 9, 2018 | 2 | USA | 20 | Neurofibroma, Plexiform | Imatinib Mesylate | Quantitative Functional Airway Response |
| NCT03433183 | 10, 2019 | 2 | USA | 21 | Malignant Peripheral Nerve Sheath Tumors NF1 |
Selumetinib Sirolimus | Clinical benefit rate of selumetinib in combination with sirolimus |
| NCT04085159 | 9, 2019 | 1/2 | China | 100 | Neurofibromatosis Schwannomatosis | Antigen-specific T cells CART/CTL and DCvac | Percentage of adverse effects |
2.5. Animal Models
3. Neurofibromatosis Type 2
3.1. Clinical Characteristics
| Bilateral vestibular schwannomas or |
| First-degree relative with neurofibromatosis type 2 plus |
| 1. Unilateral vestibular schwannomas or |
| 2. Any two of the following: Meningioma, glioma, schwannoma, or juvenile PLO |
3.2. Genetic and Molecular Characteristics
3.3. Therapeutic Strategies
3.4. Ongoing Clinical Trials
| ID | Initiation Date | Phase | Nation | N | Disease | Treatment | Primary Outcome |
|---|---|---|---|---|---|---|---|
| NCT02934256 | 7, 2016 | 2 | China | 20 | NF2 | Icotinib | Change from Baseline in volume of tumor |
| NCT02129647 | 4, 2014 | 2 | USA | 12 | NF2 Progressive VS |
Axitinib | volumetric response rates |
| NCT01345136 | 7, 2015 | 2 | USA | 4 | NF2 | RAD001, everolimus | Vestibular schwannoma volume |
| NCT01767792 | 5, 2013 | 2 | USA | 22 | NF2 Progressive VS |
Bevacizumab | Hearing |
| NCT04283669 | 2, 2020 | 2 | USA | 19 | NF2 Progressive VS |
Crizotinib | Volumetric response rate |
| NCT02831257 | 8, 2016 | 2 | USA | 18 | NF2 Meningioma |
AZD2014 | Volumetric response rate |
| NCT04374305 | 6, 2020 | 2 | USA | 80 | NF2 Vestibular Schwannoma Non-vestibular Schwannoma Meningioma Ependymoma |
Brigatinib | Volumetric response rate |
| NCT03095248 | 5, 2017 | 2 | USA | 34 | NF2 Vestibular Schwannoma Meningioma Ependymoma Glioma |
Selumetinib | Hearing response Volumetric response rate |
| NCT03079999 | 6, 2018 | 2 | USA | 300 | NF2 Vestibular schwannoma |
Aspirin | Progression-free survival |
3.5. Animal Models
4. Schwannomatosis
4.1. Clinical Characteristics
| Definite Schwannomatosis |
| A. Age >30 years and two or more schwannomas (not intradermal), at least one with histologic confirmation with no evidence of vestibular tumor on brain MRI scan and no known NF mutation |
| B. Vestibular schwannoma (pathologically confirmed) plus first-degree relative who meets the criteria of schwannomatosis |
| Possible schwannomatosis |
| A. Age <30 years plus two or more schwannomas (not intradermal), at least one with histologic confirmation with no evidence of vestibular tumor on brain MRI scan and no known NF mutation |
| B. Age >45 years plus two or more schwannomas (not dermal), at least one with histologic confirmation and no symptoms of 8th nerve dysfunction and NF type 2 |
| C. Evidence of a non-vestibular schwannoma and first-degree relative meeting criteria for definite schwannomatosis |
4.2. Genetic and Molecular Characteristics
4.3. Therapeutic Strategies
4.4. Ongoing Clinical Trials
| ID | Initiation Date | Phase | Nation | N | Disease | Treatment | Primary Outcome |
|---|---|---|---|---|---|---|---|
| NCT04163419 | 4, 2020 | 2 | USA | 46 | Schwannomatosis | Tanezumab | Change in pain level |
| NCT04085159 | 9, 2019 | 1/2 | China | 100 | Neurofibromatosis Schwannomatosis | Antigen-specific T cells CART/CTL and DCvac | Percentage of adverse effects |
References
- Coy, S.; Rashid, R.; Stemmer-Rachamimov, A.; Santagata, S. An update on the CNS manifestations of neurofibromatosis type 2. Acta Neuropathol. 2020, 139, 643–665.
- Evans, D.G.; Bowers, N.L.; Tobi, S.; Hartley, C.; Wallace, A.J.; King, A.T.; Lloyd, S.K.W.; Rutherford, S.A.; Hammerbeck-Ward, C.; Pathmanaban, O.N.; et al. Schwannomatosis: A genetic and epidemiological study. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1215.
- Nix, J.S.; Blakeley, J.; Rodriguez, F.J. An update on the central nervous system manifestations of neurofibromatosis type 1. Acta Neuropathol. 2020, 139, 625–641.
- Kresak, J.L. Neurofibromatosis: A Review of NF1, NF2, and Schwannomatosis. J. Pediatr. Genet. 2016, 5, 98–104.
- Blakeley, J.O.; Plotkin, S.R. Therapeutic advances for the tumors associated with neurofibromatosis type 1, type 2, and schwannomatosis. Neuro. Oncol. 2016, 18, 624–638.
- Campian, J.; Gutmann, D.H. CNS Tumors in Neurofibromatosis. J. Clin. Oncol. 2017, 35, 2378–2385.
- Evans, D.G.; Howard, E.; Giblin, C.; Clancy, T.; Spencer, H.; Huson, S.M.; Lallo, F. Birth incidence and prevalence of tumor-prone syndromes: Estimates from a UK family genetic register service. Am. J. Med. Genet. A 2010, 152A, 327.
- Kallionpää, R.A.; Uusitalo, E.; Leppävirta, J.; Pöyhönen, M.; Peltonen, S.; Peltonen, J. Prevalence of neurofibromatosis type 1 in the Finnish population. Genet. Med. 2018, 20, 1082.
- Ruggieri, M.; Huson, S.M. The clinical and diagnostic implications of mosaicism in the neurofibromatoses. Neurology 2001, 56, 1433.
- Legius, E.; Messiaen, L.; Wolkenstein, P.; Pancza, P.; Avery, R.A.; Berman, Y.; Blakeley, J.; Babovic-Vuksanovic, D.; Cunha, K.S.; Ferner, R.; et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet. Med. 2021, 19.
- Stephens, K.; Kayes, L.; Riccardi, V.M.; Rising, M.; Sybert, V.P.; Pagon, R.A. Preferential mutation of the neurofibromatosis type 1 gene in paternally derived chromosomes. Hum. Genet. 1992, 88, 279.
- Rasmussen, S.A.; Yang, Q.; Friedman, J.M. Mortality in neurofibromatosis 1: An analysis using U.S. death certificates. Am. J. Hum. Genet. 2001, 68, 1110–1118.
- Landry, J.P.; Schertz, K.L.; Chiang, Y.J.; Bhalla, A.D.; Yi, M.; Keung, E.Z.; Scally, C.P.; Feig, B.W.; Hunt, K.K.; Roland, C.L.; et al. Comparison of Cancer Prevalence in Patients With Neurofibromatosis Type 1 at an Academic Cancer Center vs in the General Population From 1985 to 2020. JAMA Netw. Open. 2021, 4, e210945.
- Pasmant, E.; Sabbagh, A.; Hanna, N.; Masliah-Planchon, J.; Jolly, E.; Goussard, P.; Ballerini, P.; Cartault, F.; Barbarot, S.; Landman-Parker, J.; et al. SPRED1 germline mutations caused a neurofibromatosis type 1 overlapping phenotype. J. Med. Genet. 2009, 46, 425.
- Scheffzek, K.; Ahmadian, M.R.; Wiesmüller, L.; Kabsch, W.; Stege, P.; Schmitz, F.; Wittinghofer, A. Structural analysis of the GAP-related domain from neurofibromin and its implications. EMBO J. 1998, 17, 4313–4327.
- Peltonen, S.; Kallionpää, R.A.; Peltonen, J. Neurofibromatosis type 1 (NF1) gene: Beyond café au lait spots and dermal neurofibromas. Exp. Dermatol. 2017, 26, 645–648.
- Trovó-Marqui, A.B.; Tajara, E.H. Neurofibromin: A general outlook. Clin. Genet. 2006, 70, 1–13.
- Messiaen, L.; Wimmer, K. NF1 mutational spectrum. Monogr. Hum. Genet. 2008, 16, 63.
- Kluwe, L.; Friedrich, R.E.; Peiper, M.; Friedman, J.; Mautner, V.F. Constitutional NF1 mutations in neurofibromatosis 1 patients with malignant peripheral nerve sheath tumors. Hum. Mutat. 2003, 22, 420.
- Farid, M.; Demicco, E.G.; Garcia, R.; Ahn, L.; Merola, P.R.; Cioffi, A.; Maki, R.G. Malignant peripheral nerve sheath tumors. Oncologist 2014, 19, 193–201.
- Upadhyaya, M.; Spurlock, G.; Majounie, E.; Griffiths, S.; Forrester, N.; Baser, M.; Huson, S.M.; Gareth, E.D.; Ferner, R. The heterogeneous nature of germline mutations in NF1 patients with malignant peripheral serve sheath tumours (MPNSTs). Hum. Mutat. 2006, 27, 716.
- Koczkowska, M.; Callens, T.; Gomes, A.; Sharp, A.; Chen, Y.; Hicks, A.D.; Aylsworth, A.S.; Azizi, A.A.; Basel, D.G.; Bellus, G.; et al. Expanding the clinical phenotype of individuals with a 3-bp in-frame deletion of the NF1 gene (c.2970_2972del): An update of genotype-phenotype correlation. Genet. Med. 2019, 21, 867.
- Koczkowska, M.; Chen, Y.; Callens, T.; Gomes, A.; Sharp, A.; Johnson, S.; Hsiao, M.C.; Chen, Z.; Balasubramanian, M.; Barnett, C.P.; et al. Genotype-Phenotype Correlation in NF1: Evidence for a More Severe Phenotype Associated with Missense Mutations Affecting NF1 Codons 844-848. Am. J. Hum. Genet. 2018, 102, 69.
- Koczkowska, M.; Callens, T.; Chen, Y.; Gomes, A.; Hicks, A.D.; Sharp, A.; Johns, E.; Uhas, K.A.; Armstrong, L.; Bosanko, K.A.; et al. Clinical spectrum of individuals with pathogenic NF1 missense variants affecting p.Met1149, p.Arg1276, and p.Lys1423: Genotype-phenotype study in neurofibromatosis type 1. Hum. Mutat. 2020, 41, 299.
- Quesnel, B.; Preudhomme, C.; Vanrumbeke, M.; Vachee, A.; Lai, J.L.; Fenaux, P. Absence of rearrangement of the neurofibromatosis 1 (NF1) gene in myelodysplastic syndromes and acute myeloid leukemia. Leukemia 1994, 8, 878–880.
- Messiaen, L.M.; Callens, T.; Mortier, G.; Beysen, D.; Vandenbroucke, I.; Roy, N.V.; Paepe, A.D. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum. Mutat. 2000, 15, 541.
- Castle, B.; Baser, M.E.; Huson, S.M.; Cooper, D.N.; Upadhyaya, M. Evaluation of genotype-phenotype correlations in neurofibromatosis type 1. J. Med. Genet. 2003, 40, e109.
- Nardecchia, E.; Perfetti, L.; Castiglioni, M.; Di Natale, D.; Imperatori, A.; Rotolo, N. Bullous lung disease and neurofibromatosis type-1. Monaldi Arch. Chest Dis. 2012, 77, 105.
- Katz, D.; Lazar, A.; Lev, D. Malignant peripheral nerve sheath tumour (MPNST): The clinical implications of cellular signalling pathways. Expert Rev. Mol. Med. 2009, 11, e30.
- Gross, A.M.; Wolters, P.L.; Dombi, E.; Baldwin, A.; Whitcomb, P.; Fisher, M.J.; Weiss, B.; Kim, A.; Bornhorst, M.; Shah, A.C.; et al. Selumetinib in Children with Inoperable Plexiform Neurofibromas. N. Engl. J. Med. 2020, 382, 1430–1442.
- Mukhopadhyay, S.; Maitra, A.; Choudhury, S. Selumetinib: The first ever approved drug for neurofibromatosis-1 related inoperable plexiform neurofibroma. Curr. Med. Res. Opin. 2021, 8, 1.
- DeBella, K.; Szudek, J.; Friedman, J.M. Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics 2000, 105, 608–614.
- Kaufman, L.M.; Doroftei, O. Optic glioma warranting treatment in children. Eye 2006, 20, 1149–1164.
- Brannan, C.I.; Perkins, A.S.; Vogel, K.S.; Ratner, N.; Nordlund, M.L.; Reid, S.W.; Buchberg, A.M.; Jenkins, N.A.; Parada, L.F.; Copeland, N.G. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev. 1994, 8, 1019–1029.
- Jacks, T.; Shih, T.S.; Schmitt, E.M.; Bronson, R.T.; Bernards, A.; Weinberg, R.A. Tumor predisposition in mice heterozygous for a targeted mutation in NF1. Nat. Genet. 1994, 7, 353–361.
- Lakkis, M.M.; Epstein, J.A. Neurofibromin modulation of ras activity is required for normal endocardial-mesenchymal transformation in the developing heart. Development 1998, 125, 4359–4367.
- Lakkis, M.M.; Golden, J.A.; O’Shea, S.; Epstein, J.A. Neurofibromin deficiency in mice causes exencephaly and is a modifier for Splotch neural tube defects. Dev. Biol. 1999, 212, 80–92.
- Bajenaru, M.L.; Donahoe, J.; Corral, T.; Keilly, K.M.; Brophy, S.; Pellicer, A.; Gutmann, D.H. Neurofibromatosis 1 (NF1) heterozygosity results in a cell—Autonomous growth advantage for astrocytes. Glia 2001, 33, 314–323.
- Gutmann, D.H.; Loehr, A.; Zhang, Y.; Kim, J.; Henkemeyer, M.; Cashen, A. Haploinsufficiency for the neurofibromatosis 1 (NF1) tumor suppressor results in increased astrocyte proliferation. Oncogene 1999, 18, 4450–4459.
- Zhu, Y.; Romero, M.I.; Ghosh, P.; Ye, Z.; Charnay, P.; Rushing, E.J.; Marth, J.D.; Parada, L.F. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 2001, 15, 859–876.
- Costa, R.M.; Yang, T.; Huynh, D.P.; Pulst, S.M.; Viskochil, D.H.; Silva, A.J.; Brannan, C.I. Learning deficits, but normal development and tumor predisposition, in mice lacking exon 23a of Nf1. Nat. Genet. 2001, 27, 399–405.
- Zuckermann, M.; Hovestadt, V.; Knobbe-Thomsen, C.B.; Zapatka, M.; Northcott, P.A.; Schramm, K.; Belic, J.; Jones, D.T.; Tschida, B.; Moriarity, B.; et al. Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling. Nat. Commun. 2015, 6, 7391.
- Kwon, C.H.; Zhao, D.; Chen, J.; Alcantara, S.; Li, Y.; Burns, D.K.; Mason, R.P.; Lee, E.Y.; Wu, H.; Parada, L.F. Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer Res. 2008, 68, 3286–3294.
- Reilly, K.M.; Loisel, D.A.; Bronson, R.T.; McLaughlin, M.E.; Jacks, T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat. Genet. 2000, 26, 109–113.
- Marumoto, T.; Tashiro, A.; Friedmann-Morvinski, D.; Scadeng, M.; Soda, Y.; Gage, F.H.; Verma, I.M. Development of a novel mouse glioma model using lentiviral vectors. Nat. Med. 2009, 15, 110–116.
- Bajenaru, M.L.; Hernandez, M.R.; Perry, A.; Zhu, Y.; Parada, L.F.; Garbow, J.R.; Gutmann, D.H. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res. 2003, 63, 8573–8577.
- Isakson, S.H.; Rizzardi, A.E.; Coutts, A.W.; Carlson, D.F.; Kirstein, M.N.; Fisher, J.; Vitte, J.; Williams, K.B.; Pluhar, G.E.; Dahiya, S.; et al. Genetically engineered minipigs model the major clinical features of human neurofibromatosis type 1. Commun Biol. 2018, 1, 158.
- White, K.A.; Swier, V.J.; Cain, J.T.; Kohlmeyer, J.L.; Meyerholz, D.K.; Tanas, M.R.; Uthoff, J.; Hammond, E.; Li, H.; Rohret, F.A.; et al. A porcine model of neurofibromatosis type 1 that mimics the human disease. JCI Insight. 2018, 3, e120402.
- Evans, D.G.; Hartley, C.L.; Smith, P.T.; King, A.T.; Bowers, N.L.; Tobi, S.; Wallace, A.J.; Perry, M.; Anup, R.; Lloyd, S.K.W.; et al. Incidence of mosaicism in 1055 de novo NF2 cases: Much higher than previous estimates with high utility of next-generation sequencing. Genet. Med. 2020, 22, 53.
- Evans, D.G.; Ramsden, R.T.; Shenton, A.; Gokhale, C.; Bowers, N.L.; Huson, S.M.; Pichert, G.; Wallace, A. Mosaicism in neurofibromatosis type 2: An update of risk based on uni/bilaterality of vestibular schwannoma at presentation and sensitive mutation analysis including multiple ligation-dependent probe amplification. J. Med. Genet. 2007, 44, 424.
- Evans, D.G.; Moran, A.; King, A.; Saeed, S.; Gurusinghe, N.; Ramsden, R. Incidence of vestibular schwannoma and neurofibromatosis 2 in the North West of England over a 10-year period: Higher incidence than previously thought. Otol. Neurotol. 2005, 26, 93.
- Godel, T.; Bäumer, P.; Farschtschi, S.; Gugel, I.; Kronlage, M.; Hofstadler, B.; Heiland, S.; Gelderblom, M.; Bendszus, M.; Mautner, V.F. Peripheral nervous system alterations in infant and adult neurofibromatosis type 2. Neurology 2019, 93, e590.
- Picry, A.; Bonne, N.X.; Ding, J.; Aboukais, R.; Lejeune, J.P.; Baroncini, M.; Dubrulle, F.; Vincent, C. Long-term growth rate of vestibular schwannoma in neurofibromatosis 2: A volumetric consideration. Laryngoscope 2016, 126, 2358–2362.
- Evans, D.G. Neurofibromatosis type 2 (NF2): A clinical and molecular review. Orphanet J. Rare Dis. 2009, 4, 16.
- Sperfeld, A.D.; Hein, C.; Schröder, J.M.; Ludolph, A.C.; Hanemann, C.O. Occurrence and characterization of peripheral nerve involvement in neurofibromatosis type 2. Brain 2002, 125, 996.
- Gaudioso, C.; Listernick, R.; Fisher, M.J.; Campen, C.J.; Paz, A.; Gutmann, D.H. Neurofibromatosis 2 in children presenting during the first decade of life. Neurology 2019, 93, e964.
- McLaughlin, M.E.; Pepin, S.M.; Maccollin, M.; Choopong, P.; Lessell, S. Ocular pathologic findings of neurofibromatosis type 2. Arch. Ophthalmol. 2007, 125, 389.
- Painter, S.L.; Sipkova, Z.; Emmanouil, B.; Halliday, D.; Parry, A.; Elston, J.S. Neurofibromatosis Type 2-Related Eye Disease Correlated with Genetic Severity Type. J. Neuroophthalmol. 2019, 39, 44.
- Castellanos, E.; Plana, A.; Carrato, C.; Carrió, M.; Rosas, I.; Amilibia, E.; Roca-Ribas, F.; Hostalot, C.; Castillo, A.; Ros, A.; et al. Early Genetic Diagnosis of Neurofibromatosis Type 2 from Skin Plaque Plexiform Schwannomas in Childhood. JAMA Dermatol. 2018, 154, 341.
- Kluwe, L.; Mautner, V.; Heinrich, B.; Dezube, R.; Jacoby, L.B.; Friedrich, R.E.; MacCollin, M. Molecular study of frequency of mosaicism in neurofibromatosis 2 patients with bilateral vestibular schwannomas. J. Med. Genet. 2003, 40, 109.
- Moyhuddin, A.; Baser, M.E.; Watson, C.; Purcell, S.; Ramsden, R.T.; Heiberg, A.; Wallace, A.J.; Evans, D.G. Somatic mosaicism in neurofibromatosis 2: Prevalence and risk of disease transmission to offspring. J. Med. Genet. 2003, 40, 459.
- Selvanathan, S.K.; Shenton, A.; Ferner, R.; Wallace, A.J.; Huson, S.M.; Ramsden, R.T.; Evans, D.G. Further genotype—Phenotype correlations in neurofibromatosis 2. Clin. Genet. 2010, 77, 163.
- Smith, M.J.; Higgs, J.E.; Bowers, N.L.; Halliday, D.; Paterson, J.; Gillespie, J.; Huson, S.M.; Freeman, S.R.; Lloyd, S.; Rutherford, S.A.; et al. Cranial meningiomas in 411 neurofibromatosis type 2 (NF2) patients with proven gene mutations: Clear positional effect of mutations, but absence of female severity effect on age at onset. J. Med. Genet. 2011, 48, 261.
- Hexter, A.; Jones, A.; Joe, H.; Heap, L.; Smith, M.J.; Wallace, A.J.; Halliday, D.; Parry, A.; Taylor, A.; Raymond, L.; et al. Clinical and molecular predictors of mortality in neurofibromatosis 2: A UK national analysis of 1192 patients. J. Med. Genet. 2015, 52, 699.
- Evans, D.G.; Ramsden, R.T.; Gokhale, C.; Bowers, N.; Huson, S.M.; Wallace, A. Should NF2 mutation screening be undertaken in patients with an apparently isolated vestibular schwannoma? Clin. Genet. 2007, 71, 354.
- Evans, D.G.; Wallace, A.J.; Hartley, C.; Freeman, S.R.; Lloyd, S.K.; Thomas, O.; Axon, P.; Hammerbeck-Ward, C.L.; Pathmanaban, O.; Rutherford, S.A.; et al. Familial unilateral vestibular schwannoma is rarely caused by inherited variants in the NF2 gene. Laryngoscope 2019, 129, 967.
- Evans, D.G.; Baser, M.E.; O’Reilly, B.; Rowe, J.; Gleeson, M.; Saeed, S.; King, A.; Huson, S.M.; Kerr, R.; Thomas, N.; et al. Management of the patient and family with neurofibromatosis 2: A consensus conference statement. Br. J. Neurosurg. 2005, 19, 5–12.
- Dewan, R.; Pemov, A.; Kim, H.J.; Morgan, K.L.; Vasquez, R.A.; Chittiboina, P.; Wang, X.; Chandrasekharappa, S.C.; Ray-Chaudhury, A.; Butman, J.A.; et al. Evidence of polyclonality in neurofibromatosis type 2-associated multilobulated vestibular schwannomas. Neuro. Oncol. 2015, 17, 566.
- Mathieu, D.; Kondziolka, D.; Flickinger, J.C.; Niranjan, A.; Williamson, R.; Martin, J.J.; Lunsford, L.D. Stereotactic radiosurgery for vestibular schwannomas in patients with neurofibromatosis type 2: An analysis of tumor control, complications, and hearing preservation rates. Neurosurgery 2007, 60, 460.
- Sharma, M.S.; Singh, R.; Kale, S.S.; Agrawal, D.; Sharma, B.S.; Mahapatra, A.K. Tumor control and hearing preservation after Gamma Knife radiosurgery for vestibular schwannomas in neurofibromatosis type 2. J. Neurooncol. 2010, 98, 265–270.
- McClelland, S., 3rd; Gerbi, B.J.; Cho, K.H.; Hall, W.A. The treatment of a large acoustic tumor with fractionated stereotactic radiotherapy. J. Robot. Surg. 2007, 1, 227–230.
- Seferis, C.; Torrens, M.; Paraskevopoulou, C.; Psichidis, G. Malignant transformation in vestibular schwannoma: Report of a single case, literature search, and debate. J. Neurosurg. 2014, 121, 160–166.
- Balasubramaniam, A.; Shannon, P.; Hodaie, M.; Laperriere, N.; Michaels, H.; Guha, A. Glioblastoma multiforme after stereotactic radiotherapy for acoustic neuroma: Case report and review of the literature. Neuro Oncol. 2007, 9, 447.
- Cayé-Thomasen, P. VEGF and VEGF receptor-1 concentration in vestibular schwannoma homogenates correlates to tumor growth rate. Otol. Neurotol. 2005, 26, 98–101.
- Komotar, R.J.; Starke, R.M.; Sisti, M.B.; Connolly, E.S. The role of bevacizumab in hearing preservation and tumor volume control in patients with vestibular schwannomas. Neurosurgery 2009, 65, N12.
- Alanin, M.C.; Klausen, C.; Caye-Thomasen, P.; Thomsen, C.; Fugleholm, K.; Poulsgaard, L.; Lassen, U.; Mau-Sorensen, M.; Hofland, K.F. The effect of bevacizumab on vestibular schwannoma tumour size and hearing in patients with neurofibromatosis type 2. Eur. Arch. Otorhinolaryngol. 2015, 272, 3627–3633.
- Blakeley, J.; Schreck, K.C.; Evans, D.G.; Korf, B.R.; Zagzag, D.; Karajannis, M.A.; Bergner, A.L.; Belzberg, A.J. Clinical response to bevacizumab in schwannomatosis. Neurology 2014, 83, 1986–1987.
- Eminowicz, G.K.; Raman, R.; Conibear, J.; Plowman, P.N. Bevacizumab treatment for vestibular schwannomas in neurofibromatosis type two: Report of two cases, including responses after prior gamma knife and vascular endothelial growth factor inhibition therapy. J. Laryngol. Otol. 2012, 126, 79–82.
- Liu, P.; Yao, Q.; Li, N.A.; Liu, Y.; Wang, Y.; Li, M.; Li, Z.; Li, J.; Li, G. Low-dose bevacizumab induces radiographic regression of vestibular schwannomas in neurofibromatosis type 2: A case report and literature review. Oncol. Lett. 2016, 11, 2981–2986.
- Mautner, V.F.; Nguyen, R.; Kutta, H.; Fuensterer, C.; Bokemeyer, C.; Hagel, C.; Friedrich, R.E.; Panse, J. Bevacizumab induces regression of vestibular schwannomas in patients with neurofibromatosis type 2. Neuro. Oncol. 2010, 12, 14–18.
- Plotkin, S.R.; Stemmer-Rachamimov, A.O.; Barker, F.G., 2nd; Halpin, C.; Padera, T.P.; Tyrrell, A.; Sorensen, A.G.; Jain, R.K.; di Tomaso, E. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N. Engl. J. Med. 2009, 361, 358–367.
- Plotkin, S.R.; Merker, V.L.; Halpin, C.; Jennings, D.; McKenna, M.J.; Harris, G.J.; Barker, F.G., 2nd. Bevacizumab for progressive vestibular schwannoma in neurofibromatosis type 2: A retrospective review of 31 patients. Otol. Neurotol. 2012, 33, 1046–1052.
- Subbiah, V.; Slopis, J.; Hong, D.S.; Ketonen, L.M.; Hamilton, J.; McCutcheon, I.E.; Kurzrock, R. Treatment of patients with advanced neurofibromatosis type 2 with novel molecularly targeted therapies: From bench to bedside. J. Clin. Oncol. 2012, 30, 64–68.
- Versleijen, M.W.; Verbist, B.M.; Mulder, J.J.; de Geus-Oei, L.F.; van Herpen, C.M. Avastin scintigraphy in surveillance of bevacizumab treatment in a patient with neurofibromatosis type 2: A case report. Clin. Nucl. Med. 2014, 39, 277–280.
- Morris, K.A.; Golding, J.F.; Axon, P.R.; Afridi, S.; Blesing, C.; Ferner, R.E.; Halliday, D.; Jena, R.; Pretorius, P.M. Bevacizumab in Neurofibromatosis type 2 (NF2) related vestibular schwannomas: A nationally coordinated approach to delivery and prospective evaluation. Neuro-Oncol. Pract. 2016, 3, 281–289.
- Lu, V.M.; Ravindran, K.; Graffeo, C.S.; Perry, A.; Van Gompel, J.J.; Daniels, D.J.; Link, M.J. Efficacy and safety of bevacizumab for vestibular schwannoma in neurofibromatosis type 2: A systematic review and meta-analysis of treatment outcomes. J. Neurooncol. 2019, 144, 239.
- Plotkin, S.R.; Duda, D.G.; Muzikansky, A.; Allen, J.; Blakeley, J.; Rosser, T.; Campian, J.L.; Clapp, D.W.; Fisher, M.J.; Tonsgard, J.; et al. Multicenter, Prospective, Phase II and Biomarker Study of High-Dose Bevacizumab as Induction Therapy in Patients with Neurofibromatosis Type 2 and Progressive Vestibular Schwannoma. J. Clin. Oncol. 2019, 37, 3446.
- Ouerdani, A.; Goutagny, S.; Kalamarides, M.; Trocóniz, I.F.; Ribba, B. Mechanism-based modeling of the clinical effects of bevacizumab and everolimus on vestibular schwannomas of patients with neurofibromatosis type 2. Cancer Chemother. Pharmacol. 2016, 77, 1263–1273.
- Tamura, R.; Fujioka, M.; Morimoto, Y.; Ohara, K.; Kosugi, K.; Oishi, Y.; Sato, M.; Ueda, R.; Fujiwara, H.; Hikichi, T.; et al. A VEGF receptor vaccine demonstrates preliminary efficacy in neurofibromatosis type 2. Nat. Commun. 2020, 11, 2028.
- Goutagny, S.; Raymond, E.; Esposito-Farese, M.; Trunet, S.; Mawrin, C.; Bernardeschi, D.; Larroque, B.; Sterkers, O.; Giovannini, M.; Kalamarides, M. Phase II study of mTORC1 inhibition by everolimus in neurofibromatosis type 2 patients with growing vestibular schwannomas. J. Neurooncol. 2015, 122, 313.
- Karajannis, M.A.; Legault, G.; Hagiwara, M.; Giancotti, F.G.; Filatov, A.; Derman, A.; Hochman, T.; Goldberg, J.D.; Vega, E.; Wisoff, J.H.; et al. Phase II study of everolimus in children and adults with neurofibromatosis type 2 and progressive vestibular schwannomas. Neuro. Oncol. 2014, 16, 292.
- Karajannis, M.A.; Legault, G.; Hagiwara, M.; Ballas, M.S.; Brown, K.; Nusbaum, A.O.; Hochman, T.; Goldberg, J.D.; Koch, K.M.; Golfinos, J.G.; et al. Phase II trial of lapatinib in adult and pediatric patients with neurofibromatosis type 2 and progressive vestibular schwannomas. Neuro. Oncol. 2012, 14, 1163.
- Plotkin, S.R.; Halpin, C.; McKenna, M.J.; Loeffler, J.S.; Batchelor, T.T.; Barker, F.G., 2nd. Erlotinib for progressive vestibular schwannoma in neurofibromatosis 2 patients. Otol. Neurotol. 2010, 31, 1135.
- Tamura, R.; Morimoto, Y.; Sato, M.; Kuranari, Y.; Oishi, Y.; Kosugi, K.; Yoshida, K.; Toda, M. Difference in the hypoxic immunosuppressive microenvironment of patients with neurofibromatosis type 2 schwannomas and sporadic schwannomas. J. Neurooncol. 2020, 146, 265–273.
- Deep, N.L.; Patel, E.J.; Shapiro, W.H.; Waltzman, S.B.; Jethanamest, D.; McMenomey, S.O.; Roland, J.T., Jr.; Friedmann, D.R. Cochlear Implant Outcomes in Neurofibromatosis Type 2: Implications for Management. Otol. Neurotol. 2021, 42, 540–548.
- Neff, B.A.; Wiet, R.M.; Lasak, J.M.; Cohen, N.L.; Pillsbury, H.C.; Ramsden, R.T.; Welling, D.B. Cochlear implantation in the neurofibromatosis type 2 patient: Long-term follow-up. Laryngoscope 2007, 117, 1069.
- Funes, C.J.; Mace, R.A.; Macklin, E.A.; Plotkin, S.R.; Jordan, J.T.; Vranceanu, A.M. First report of quality of life in adults with neurofibromatosis 2 who are deafened or have significant hearing loss: Results of a live-video randomized control trial. J. Neurooncol. 2019, 143, 505.
- Evans, D.G.; Watson, C.; King, A.; Wallace, A.J.; Baser, M.E. Multiple meningiomas: Differential involvement of the NF2 gene in children and adults. J. Med. Genet. 2005, 42, 45.
- Larson, J.J.; van Loveren, H.R.; Balko, M.G.; Tew, J.M., Jr. Evidence of meningioma infiltration into cranial nerves: Clinical implications for cavernous sinus meningiomas. J. Neurosurg. 1995, 83, 596.
- Wentworth, S.; Pinn, M.; Bourland, J.D.; Deguzman, A.F.; Ekstrand, K.; Ellis, T.L.; Glazier, S.S.; McMullen, K.P.; Munley, M.; Stieber, V.W.; et al. Clinical experience with radiation therapy in the management of neurofibromatosis-associated central nervous system tumors. Int. J. Radiat. Oncol. Biol. Phys. 2009, 73, 208.
- Osorio, D.S.; Hu, J.; Mitchell, C.; Allen, J.C.; Stanek, J.; Hagiwara, M.; Karajannis, M.A. Effect of lapatinib on meningioma growth in adults with neurofibromatosis type 2. J. Neurooncol. 2018, 139, 749.
- Nunes, F.P.; Merker, V.L.; Jennings, D.; Caruso, P.A.; di Tomaso, E.; Muzikansky, A.; Barker, F.G., 2nd; Stemmer-Rachamimov, A.; Plotkin, S.R. Bevacizumab treatment for meningiomas in NF2: A retrospective analysis of 15 patients. PLoS ONE 2013, 8, e59941.
- Fujii, M.; Ichikawa, M.; Iwatate, K.; Bakhit, M.; Yamada, M.; Kuromi, Y.; Sato, T.; Sakuma, J.; Saito, K. Bevacizumab Therapy of Neurofibromatosis Type 2 Associated Vestibular Schwannoma in Japanese Patients. Neurol. Med. Chir. 2020, 60, 75–82.
- McClatchey, A.I.; Saotome, I.; Ramesh, V.; Gusella, J.F.; Jacks, T. The NF2 tumor suppressor gene product is essential for extraembryonic development immediately prior to gastrulation. Genes Dev. 1997, 11, 1253–1265.
- Giovannini, M.; Robanus-Maandag, E.; Niwa-Kawakita, M.; van der Valk, M.; Woodruff, J.M.; Goutebroze, L.; Merel, P.; Berns, A.; Thomas, G. Schwann cell hyperplasia and tumors in transgenic mice expressing a naturally occurring mutant NF2 protein. Genes Dev. 1999, 13, 978–986.
- Kalamarides, M.; Niwa-Kawakita, M.; Leblois, H.; Abramowski, V.; Perricaudet, M.; Janin, A.; Thomas, G.; Gutmann, D.H.; Giovanni, M. Nf2 gene inactivation in arachnoidal cells is rate-limiting for meningioma development in the mouse. Genes Dev. 2002, 16, 1060–1065.
- Chen, J.; Landegger, L.D.; Sun, Y.; Ren, J.; Maimon, N.; Wu, L.; Ng, M.R.; Chen, J.W.; Zhang, N.; Zhao, Y.; et al. A cerebellopontine angle mouse model for the investigation of tumor biology, hearing, and neurological function in NF2-related vestibular schwannoma. Nat. Protoc. 2019, 14, 541–555.
- Kalamarides, M.; Stemmer-Rachamimov, A.O.; Takahashi, M.; Han, Z.Y.; Chareyre, F.; Niwa-Kawakita, M.; Black, P.M.; Carroll, R.S.; Giovannini, M. Natural history of meningioma development in mice reveals: A synergy of Nf2 and p16(Ink4a) mutations. Brain Pathol. 2008, 18, 62–70.
- Burns, S.S.; Chang, L.S. Generation of noninvasive, quantifiable, orthotopic animal models for NF2-associated schwannoma and meningioma. Methods Mol. Biol. 2016, 1427, 59–72.
- MacCollin, M.; Woodfin, W.; Kronn, D.; Short, M.P. Schwannomatosis: A clinical and pathologic study. Neurology 1996, 46, 1072.
- Plotkin, S.R.; Bredella, M.A.; Cai, W.; Kassarjian, A.; Harris, G.J.; Esparza, S.; Merker, V.L.; Munn, L.L.; Muzikansky, A.; Askenazi, M.; et al. Quantitative assessment of whole-body tumor burden in adult patients with neurofibromatosis. PLoS ONE 2012, 7, e35711.
- Merker, V.L.; Esparza, S.; Smith, M.J.; Stemmer-Rachamimov, A.; Plotkin, S.R. Clinical features of schwannomatosis: A retrospective analysis of 87 patients. Oncologist 2012, 17, 1317.
- MacCollin, M.; Chiocca, E.A.; Evans, D.G.; Friedman, J.M.; Horvitz, R.; Jaramillo, D.; Lev, M.; Mautner, V.F.; Niimura, M.; Plotkin, S.R.; et al. Diagnostic criteria for schwannomatosis. Neurology 2005, 64, 1838.
- Carter, J.M.; O’Hara, C.; Dundas, G.; Gilchrist, D.; Collins, M.S.; Eaton, K.; Judkins, A.R.; Biegel, J.A.; Folpe, A.L. Epithelioid malignant peripheral nerve sheath tumor arising in a schwannoma, in a patient with “neuroblastoma-like” schwannomatosis and a novel germline SMARCB1 mutation. Am. J. Surg. Pathol. 2012, 36, 154.
- Jacoby, L.B.; MacCollin, M.; Parry, D.M.; Kluwe, L.; Lynch, J.; Jones, D.; Gusella, J.F. Allelic expression of the NF2 gene in neurofibromatosis 2 and schwannomatosis. Neurogenetics 1999, 2, 101.
- Van den Munckhof, P.; Christiaans, I.; Kenter, S.B.; Baas, F.; Hulsebos, T.J. Germline SMARCB1 mutation predisposes to multiple meningiomas and schwannomas with preferential location of cranial meningiomas at the falx cerebri. Neurogenetics 2012, 13, 1.
- Hadfield, K.D.; Newman, W.G.; Bowers, N.L.; Wallace, A.; Bolger, C.; Colley, A.; McCann, E.; Trump, D.; Prescott, T.; Evans, D.G. Molecular characterisation of SMARCB1 and NF2 in familial and sporadic schwannomatosis. J. Med. Genet. 2008, 45, 332.
- Boyd, C.; Smith, M.J.; Kluwe, L.; Balogh, A.; Maccollin, M.; Plotkin, S.R. Alterations in the SMARCB1 (INI1) tumor suppressor gene in familial schwannomatosis. Clin. Genet. 2008, 74, 358.
- Hadfield, K.D.; Smith, M.J.; Urquhart, J.E.; Wallace, A.J.; Bowers, N.L.; King, A.T.; Rutherford, S.A.; Trump, D.; Newman, W.G.; Evans, D.G. Rates of loss of heterozygosity and mitotic recombination in NF2 schwannomas, sporadic vestibular schwannomas and schwannomatosis schwannomas. Oncogene 2010, 29, 6216.
- Hulsebos, T.J.; Kenter, S.B.; Jakobs, M.E.; Baas, F.; Chong, B.; Delatycki, M.B. SMARCB1/INI1 maternal germ line mosaicism in schwannomatosis. Clin. Genet. 2010, 77, 86.
- Sestini, R.; Bacci, C.; Provenzano, A.; Genuardi, M.; Papi, L. Evidence of a four-hit mechanism involving SMARCB1 and NF2 in schwannomatosis-associated schwannomas. Hum. Mutat. 2008, 29, 227.
- Rousseau, G.; Noguchi, T.; Bourdon, V.; Sobol, H.; Olschwang, S. SMARCB1/INI1 germline mutations contribute to 10% of sporadic schwannomatosis. BMC Neurol. 2011, 11, 9.
- Smith, M.J.; Wallace, A.J.; Bowers, N.L.; Rustad, C.F.; Woods, C.G.; Leschziner, G.D.; Ferner, R.E.; Evans, D.G. Frequency of SMARCB1 mutations in familial and sporadic schwannomatosis. Neurogenetics 2012, 13, 141.
- Plotkin, S.R.; Blakeley, J.O.; Evans, D.G.; Hanemann, C.O.; Hulsebos, T.J.; Hunter-Schaedle, K.; Kalpana, G.V.; Korf, B.; Messiaen, L.; Papi, L.; et al. Update from the 2011 International Schwannomatosis Workshop: From genetics to diagnostic criteria. Am. J. Med. Genet. A 2013, 161A, 405.
- Versteege, I.; Sévenet, N.; Lange, J.; Rousseau-Merck, M.F.; Ambros, P.; Handgretinger, R.; Aurias, A.; Delattre, O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998, 394, 203.
- Eaton, K.W.; Tooke, L.S.; Wainwright, L.M.; Judkins, A.R.; Biegel, J.A. Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr. Blood Cancer 2011, 56, 7.
- Swensen, J.J.; Keyser, J.; Coffin, C.M.; Biegel, J.A.; Viskochil, D.H.; Williams, M.S. Familial occurrence of schwannomas and malignant rhabdoid tumour associated with a duplication in SMARCB1. J. Med. Genet. 2009, 46, 68.
- Caltabiano, R.; Magro, G.; Polizzi, A.; Praticò, A.D.; Ortensi, A.; D’Orazi, V.; Panunzi, A.; Milone, P.; Maiolino, L.; Nicita, F.; et al. A mosaic pattern of INI1/SMARCB1 protein expression distinguishes Schwannomatosis and NF2-associated peripheral schwannomas from solitary peripheral schwannomas and NF2-associated vestibular schwannomas. Childs Nerv. Syst. 2017, 33, 933.
- Piotrowski, A.; Xie, J.; Liu, Y.F.; Poplawski, A.B.; Gomes, A.R.; Madanecki, P.; Fu, C.; Crowley, M.R.; Crossman, D.K.; Armstrong, L.; et al. Germline loss-of-function mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas. Nat. Genet. 2014, 46, 182.
- Smith, M.J.; Isidor, B.; Beetz, C.; Williams, S.G.; Bhaskar, S.S.; Richer, W.; O’Sullivan, J.; Anderson, B.; Daly, S.B.; Urquhart, J.E.; et al. Mutations in LZTR1 add to the complex heterogeneity of schwannomatosis. Neurology 2015, 84, 141.
- Frattini, V.; Trifonov, V.; Chan, J.M.; Castano, A.; Lia, M.; Abate, F.; Keir, S.T.; Ji, A.X.; Zoppoli, P.; Niola, F.; et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat. Genet. 2013, 5, 1141.
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011, 39, D945.
- Algar, E.M.; Muscat, A.; Dagar, V.; Rickert, C.; Chow, C.W.; Biegel, J.A.; Ekert, P.G.; Saffery, R.; Craig, J.; Johnstone, R.W.; et al. Imprinted CDKN1C is a tumor suppressor in rhabdoid tumor and activated by restoration of SMARCB1 and histone deacetylase inhibitors. PLoS ONE 2009, 4, e4482.
- Emanuele, M.J.; Elia, A.E.; Xu, Q.; Thoma, C.R.; Izhar, L.; Leng, Y.; Guo, A.; Chen, Y.N.; Rush, J.; Hsu, P.W.; et al. Global identification of modular cullin-RING ligase substrates. Cell 2011, 147, 459.


