 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Feifei Guo | + 2264 word(s) | 2264 | 2021-04-27 05:37:48 | | | |
| 2 | Dean Liu | -10 word(s) | 2254 | 2021-06-02 05:42:26 | | |
Video Upload Options
Chimeric antigen receptor (CAR)-T cell therapy has exhibited good application prospects in the treatment of hematologic malignancies. However, there are still many unsolved problems, such as the limited antitumor effect of CAR-T on solid tumors and the potential risk of CAR-T therapy in clinical applications. In order to meet these challenges, more and more solutions are proposed.
1. Introduction
In 2017, chimeric antigen receptor (CAR)-T cell products, represented by Kymriah (CTL019) [1] and Yescarta (KTE-C19) [2], were approved for marketing by the Food and Drug Administration (FDA), which highlights the therapeutic potential of CAR-T in hematologic malignancies. However, despite being a promising antitumor candidate, CAR-T still faces many problems as it continues to encounter sequent obstacles in the treatment of solid tumors. It is difficult for CAR-T cells to properly migrate and infiltrate into solid tumor sites due to the chemokine mismatches between CAR-T and tumor cells [3] and the presence of abnormal vascular [4] and stromal [5] components in the tumor microenvironment (TME). Subsequently, CAR-T cells entering tumor tissues compete with diverse immunosuppressive cells (e.g., regulatory T cells and myeloid-derived suppressor cells) [6][7], immune checkpoints (e.g., programmed death-ligand 1 [PD-L1]) [8], and metabolic checkpoints (e.g., hypoxia and glycolysis) [9][10] in the TME. Even after successfully entering tumor tissues, these CAR-T cells have to cope with problems such as tumor antigen heterogeneity and antigen escape [11][12]. Moreover, activity reduction and persistence limitation of infused CAR-T products may be the leading cause of tumor recurrence in hematologic and solid tumors [13]. The inevitable immune-mediated toxicity, manifested as cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS), is also a critical problem for the clinical application of CAR-T [14]. Although mild patients may present only self-limited symptoms such as fever and hypotension, life-threatening organ damage and death can also occur, similar to that reported in the P-PSMA-101 study or with the UCARTCS1 product [15]. Although CAR-T products of good manufacturing practice (GMP) grade are currently available, the CAR-T manufacturing process still needs to be improved to increase clinical access [16]. Methods of obtaining T cell isolation, activation, expansion, and genetic modification more effectively and economically are worth exploring [17][18]. The success of CAR-T therapy in cancer treatment traps it into obstacles. Recently, more effective solutions have been developed and reported. Therefore, in this review, we have discussed and summarized the recent remarkable improvements and breakthroughs in CAR-T therapy to address the existing challenges (Figure 1).
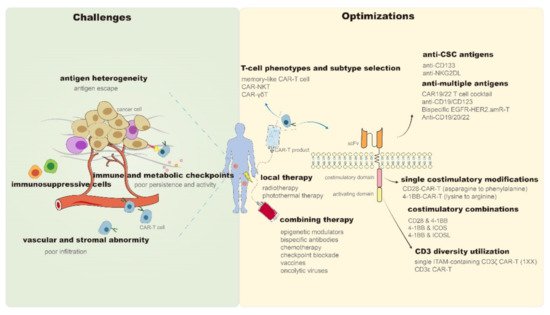
Figure 1. Challenges and optimizations of CAR-T persistence and antitumor properties. CAR—Chimeric antigen receptor; NKT—Natural killer T; γδT—gamma delta T; CSC—Cancer stem cells; CD—Cluster of differentiation; NKG2DL—Natural killer group 2 member D ligand; EGFR—Epidermal growth factor receptor; HER2—Human epidermal growth factor receptor 2; amR—Antibody mimic receptor; scFv—Single-chain variable fragment; 4-1BB (TNFRSF9)—Tumor necrosis factor receptor superfamily 9; ICOS—Combining inducible costimulatory; ICOSL—Combining inducible costimulatory ligand; ITAM—Immunoreceptor tyrosine-based activation motif.
2. Enhanced Clinical Safety and Accessibility of CAR-T Therapy
2.1. Safety Maintenance of CAR-T Therapy
Adverse effects (AEs) are a major impediment to the effective application of CAR-T therapy. Life-threatening toxicity and consequent death or treatment abandonment of patients can be commonly seen in clinical trials [19]. Just like what Penack et al. have reviewed, the unique toxicity profile of CAR-T, including CRS, ICANS, cardiotoxicity, pulmonary toxicity, metabolic complications, secondary macrophage-activation syndrome (sHLH/MAS), and prolonged cytopenia, are also frequently occurring events in clinical-trial widely utilized CD19 CAR-T therapy [20]. Additionally, the personalized responses of cancer patients to CAR-T toxicity make it a more serious problem [21]. CRS, as the most common side effect of CAR-T, has characteristic clinical symptoms ranging from mild (e.g., flu-like fever, fatigue, and headache) to severe (e.g., life-threatening multi-organ system failure). Additionally, the positive feedback loop between tumor-antigen binding CAR-T produced inflammatory cytokines (e.g., TNF-α, IFN-γ) and subsequently activated bystander immune cell (e.g., macrophages and endothelial cells) released proinflammatory cytokines (e.g., IL-1 and IL-6) is considered as the main mechanism of CRS occurrence [22]. ICANS is the second most common side effect in CAR-T therapy along with CRS or subsequently, which has a wide range of clinical manifestations, such as language/behavioral disturbance, peripheral neuropathy, and acute cerebral edema [23]. Although the pathogenesis of ICANS has not been fully understood, blood–brain barrier (BBB) disruption after high-concentration proinflammatory cytokine induced endothelial activation and the diffusion of both CAR-T and cytokines into the central nervous system are thought to be the key contributors [24]. To solve these problems, many management strategies, such as accurate grading and intervention, have been adopted to minimize CAR-T toxicities [25]. Currently an increasing number of safety switches that can directly prevent or block CAR-T toxicities have been developed to help avoid AEs in clinical cancer patients without weakening CAR-T efficacy, and remarkable results were observed both preclinically and clinically [26].
2.1.1. The Endogenous Witches
Inhibitory CAR (iCAR), comprised of an scFv targeting normal tissue-specific antigens and a powerful acute inhibitory signaling domain of immunoinhibitory receptors, is a promising endogenous approach to restrict unwanted AEs in CAR-T therapy. Fedorov et al. constructed a PD-1 and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)-based iCAR-T and confirmed its selective and reversible suppression of T cell activity, which provides a dynamic safety switch to prevent potential on-target off-tumor effects [27]. Another example is the killer cell immunoglobulin-like receptor (KIR)/PD-1-based iCAR (iKP CAR). The anti-CD19 CAR-T integrated with iKP CAR not only exhibited significant cytotoxicity to malignant B cells but also averted the damage of CD19-positive healthy B cells in vitro and in Burkitt’s lymphoma xenograft models [28].
The synthetic Notch (SynNotch) system is another typical endogenous safety switch. When the SynNotch part is activated by one antigen on tumor cells, the transcription of a CAR structure that recognizes a second tumor antigen is initiated [29], which allows for a specific activation of these dual-receptor AND-gate CAR-T cells by dual-antigen tumor cells [30]. In response to fatal bone marrow (containing ROR1+ stromal cells) failure caused by ROR1 specific CAR-T in mouse models, Srivastava et al. designed a CAR-T with a EpCAM or B7-H3 (antigens expressed on ROR1+ tumor cells but not ROR1+ stromal cells) specific synNotch receptor to enhance selectivity, which permitted both the safe targeting of tumor cells sufficiently separated from normal cells and the prevention of potential on-target off-tumor effects [31].
2.1.2. The Exogenous Switches
Small-molecule drugs are the most common choice for exogenous safety switches because they can regulate cellular function by modulating protein–protein interactions. Safety switches can be divided into on and off types according to their functions, both of which have been applied to ensure CAR-T safety. Regarding the on-switches, lipocalin-based on-switch has been reported to be a suitable option for CAR-T as the human retinol binding protein 4 (hRBP4, a member of the lipocalin family) interacts with engineered hRBP4 binders in the presence of the small molecule A1120 and, thus, successfully regulated the activity of CAR-T cells [32]. Regarding the off-switches, Giordano-Attianese et al. developed a STOP-CAR-T by incorporating a chemically disruptable heterodimer (CDH) into a synthetic heterodimeric CAR. CDH has a high-affinity protein interface and can be disrupted by small-molecule drugs, which allows for the dynamic inactivation of CAR-T under timed administration [33]. Another example is an Fms-related tyrosine kinase 3 (FLT3)-specific CAR-T engineered with a rituximab-responsive off-switch, which effectively guaranteed the recovery of bone marrow after AML remission [34].
A suicide gene is also a popular choice for safety switches. The inducible caspase 9 (iCasp9) is a well-known suicide gene that has been incorporated into CAR-T cells to eliminate inappropriately activated CAR-T cells. Gargett et al. demonstrated the application of AP1903 (a small-molecule dimerizer drug) in triggering rapid apoptosis of CAR-T cells that are highly expressed by iCasp9 [35]. Another suicide gene is the herpes simplex virus type 1 thymidine kinase (HSV1-tk) gene. It has been found to be capable of inducing the suicide of transduced CAR-T cells under the administration of the prodrug ganciclovir (GCV). Incorporating CAR-T with a mutated-version HSV1-tk gene (sr39tk), Murty et al. constructed a B7H3-specific sr39tk CAR-T and confirmed its complete ablation in osteosarcoma models after the intraperitoneal administration of GCV by bioluminescence and positron emission tomography (PET) imaging [36].
In addition to these irreversible all-or-none switches, an increasing number of adjustable switches have also been devised to acquire a more flexible and accurate modulation of CAR-T activity. Through the fusion of CAR into the ligand-induced degradation (LID) domain, Richman et al. created a novel CAR-LID structure and confirmed its ability to downregulate CAR expression on demand in the presence of small-molecule ligands in CAR-T-treated tumor models. This is due to the small-molecule ligand-induced exposure of a cryptic degron within LID, which can further lead to the degradation of CAR-LID protein and the loss of CAR expression on T cells [37]. Peptide-specific switchable CAR-T (sCAR-T) is another promising choice. The bifunctional switch, consisting of a tumor antigen-specific Fab and a peptide neo-epitope, can exclusively bind to sCAR-T and regulate its activity in a dose-dependent manner. The dynamic activity modulation of CD19-specific sCAR-T in B cell leukemia xenograft models is a successful example [38]. The dose-titratable administration of the tyrosine kinase inhibitor dasatinib can also achieve dynamic regulation of CAR-T function. It can induce a function-off state in the CAR structure by restraining the phosphorylation of CD3ζ and the ζ-chain of T cell receptor-associated protein kinase 70 kDa (ZAP70), with complete recovery after discontinuation. A protective effect was confirmed in a CAR-T-treated CRS mouse model [39]. Based on the continuously updated design of safety switches, many platforms have been established to make CAR-T therapy more reliable, such as the avidity-controlled CAR platform (integrating inducible and controllable functions into CAR structures) [40], the humanized artificial receptor platform (steering CAR-T by bispecific targeting molecules) [41], and different universal CAR platforms [42][43][44].
2.2. Accessibility Increase of CAR-T Therapy
2.2.1. Acquisition of T Cells
Currently, autologous T cells are the main source of CAR-T cells. However, the high cost and complicated acquisition process and the limited availability of T cells from immunosuppressive or immunodeficient cancer patients restrict the wide clinical application of autologous CAR-T therapy [45]. Developing techniques with both high efficiency and low cost to separate T cells is one of the solutions. DNA aptamers, for example, have been reported to be effective tools to achieve high-purity T cell isolation. Kacherovsky et al. have confirmed that DNA aptamers can help isolate pure and traceless CD8-T cells at low cost and with high yield; CAR-T cells derived from these cells were comparable to antibody-isolated CAR-T cells in terms of proliferation, phenotype, and efficacy in B cell lymphoma mouse models [46]. Exploration of allogeneic universal CAR-T cells is another potential strategy [47]. Peripheral blood mononuclear cells (PBMCs) from healthy donors, umbilical cord blood (UCB), and induced pluripotent stem cells (iPSCs) are currently the main allogeneic sources [48]. Among these, FT819, an iPSC-derived off-the-shelf CAR-T product, has emerged as a promising option [49]. It is a novel CD19 CAR-T designed by Fate Therapeutics, which encodes CAR with a single immunoreceptor tyrosine-based activation motif (1XX) to balance the effector and memory programs of CAR-T [50]. It was inserted into the T cell receptor alpha constant (TRAC) locus to delay the differentiation and exhaustion of CAR-T [51] and edited for TCR-expression elimination through the bi-allelic disruption of TRAC to mitigate the risk of GvHD [52]. According to a report at the 2019 ASH annual meeting, FT819 CAR-T has exhibited superior antitumor efficacy and survival compared to primary CD19 CAR-T in pre-clinical models, providing support for the subsequent first-of-kind phase I clinical trial [53].
2.2.2. Optimization of CAR-T Manufacturing Process
Currently, viral vector-mediated semi-random DNA integration is the main approach for expressing CAR structures on T cells [54]. However, the complex and expensive manufacturing processes of viral vectors, cumbersome production, quality control steps of viral vector-transfected CAR-T, and inevitable long-term monitoring of unanticipated side effects also limit its wide clinical application. In view of this, non-viral delivery systems with low-cost and high-safety properties have emerged as a better choice [55].
PiggyBac (PB) and Sleeping Beauty (SB) transposon systems, CRISPR system (clustered regularly interspaced short palindromic repeat)-mediated site-directed integration, and mRNA carriers are all popular non-viral methods with confirmed effectiveness in CAR-T transfection; the latest improvement in each has further optimized their efficacy in CAR-T manufacturing. For PB, Bishop et al. reported that dbDNA, a minimal DNA vector lacking undesirable plasmid features, may be a viable alternative in PB-mediated CAR-T generation with lower risks and costs for clinical application [56]. Meanwhile, Querques et al. constructed a high-solubility SB transposase (hsSB) based on the crystal structure of the hyperactive SB100X variant to overcome uncontrolled transposase activity and further confirmed that hsSB could generate CAR-T cells with superior antitumor efficacy in the absence of transfection reagents [57]. CRISPR-Cas9-mediated CAR-T construction with endogenous gene disruption is another optimization. For example, a CD19 CAR-T integrating CAR to the PD1 locus not only exhibited significant antitumor efficacy in pre-clinical models but also presented safety and effectiveness in r/r aggressive B-cell non-Hodgkin lymphoma (B-NHL) patients in investigator-initiated clinical trials (IITs) [58]. Moreover, compared to the Cas9-based approach, the AAV-Cpf1 system (combining adeno-associated virus with CRISPR-Cpf1) has been reported to be more efficient in generating double knockings in CAR-T cells. An example is CD22-specific AAV-Cpf1 KIKO CAR-T, a CAR-T product with homology-directed repair knockin and immune checkpoint knockout, which has exhibited lower-level exhaustion markers but comparable tumor control effect with regard to Cas9 CAR-T [59]. Targeted mRNA nanocarriers are absorbent owing to their simplicity. By simply mixing them with lymphocytes, these mRNA nanocarriers can efficiently mediate the genome editing of CAR-T cells, during which the disruption of TCR or the transformation of the central memory phenotype can also be achieved [60].
References
- Sheridan, C. First approval in sight for Novartis’ CAR-T therapy after panel vote. Nat. Biotechnol. 2017, 35, 691–693.
- Locke, F.L.; Go, W.Y.; Neelapu, S.S. Development and Use of the Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy Axicabtagene Ciloleucel in Large B-Cell Lymphoma A Review. JAMA Oncol. 2020, 6, 281–290.
- Slaney, C.Y.; Kershaw, M.H.; Darcy, P.K. Trafficking of T Cells into Tumors. Cancer Res. 2014, 74, 7168–7174.
- Baluk, P.; Hashizume, H.; McDonald, D.M. Cellular abnormalities of blood vessels as targets in cancer. Curr. Opin. Genet. Dev. 2005, 15, 102–111.
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2020, 6, 160.
- Burga, R.A.; Thorn, M.; Point, G.R.; Guha, P.; Nguyen, C.T.; Licata, L.A.; DeMatteo, R.P.; Ayala, A.; Espat, N.J.; Junghans, R.P.; et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol. Immunother. 2015, 64, 817–829.
- Budhu, S.; Schaer, D.A.; Li, Y.; Toledo-Crow, R.; Panageas, K.; Yang, X.; Zhong, H.; Houghton, A.N.; Silverstein, S.C.; Merghoub, T.; et al. Blockade of surface-bound TGF-beta on regulatory T cells abrogates suppression of effector T cell function in the tumor microenvironment. Sci. Signal. 2017, 10, eaak9702.
- Hu, W.; Zi, Z.; Jin, Y.; Li, G.; Shao, K.; Cai, Q.; Ma, X.; Wei, F. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol. Immunother. 2019, 68, 365–377.
- Berahovich, R.; Liu, X.; Zhou, H.; Tsadik, E.; Xu, S.; Golubovskaya, V.; Wu, L. Hypoxia Selectively Impairs CAR-T Cells in vitro. Cancers 2019, 11, 602.
- Cascone, T.; McKenzie, J.A.; Mbofung, R.M.; Punt, S.; Wang, Z.; Xu, C.; Williams, L.J.; Wang, Z.; Bristow, C.A.; Carugo, A.; et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab. 2018, 27, 977–987.
- Chen, N.; Li, X.; Chintala, N.K.; Tano, Z.E.; Adusumilli, P.S. Driving CARs on the uneven road of antigen heterogeneity in solid tumors. Curr. Opin. Immunol. 2018, 51, 103–110.
- Anurathapan, U.; Chan, R.C.; Hindi, H.F.; Mucharla, R.; Bajgain, P.; Hayes, B.C.; Fisher, W.E.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; et al. Kinetics of Tumor Destruction by Chimeric Antigen Receptor-modified T Cells. Mol. Ther. 2014, 22, 623–633.
- Lesch, S.; Benmebarek, M.-R.; Cadilha, B.L.; Stoiber, S.; Subklewe, M.; Endres, S.; Kobold, S. Determinants of response and resistance to CAR T cell therapy. Semin. Cancer Biol. 2020, 65, 80–90.
- Chou, C.K.; Turtle, C.J. Insight into mechanisms associated with cytokine release syndrome and neurotoxicity after CD19 CAR-T cell immunotherapy. Bone Marrow Transplant. 2019, 54, 780–784.
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851.
- Poorebrahim, M.; Sadeghi, S.; Fakhr, E.; Abazari, M.F.; Poortahmasebi, V.; Kheirollahi, A.; Askari, H.; Rajabzadeh, A.; Rastegarpanah, M.; Linē, A.; et al. Production of CAR T-cells by GMP-grade lentiviral vectors: Latest advances and future prospects. Crit. Rev. Clin. Lab. Sci. 2019, 56, 393–419.
- Gee, A.P. GMP CAR-T cell production. Best Pract. Res. Clin. Haematol. 2018, 31, 126–134.
- Dai, X.; Mei, Y.; Cai, D.; Han, W. Standardizing CAR-T therapy: Getting it scaled up. Biotechnol. Adv. 2019, 37, 239–245.
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62.
- Penack, O.; Koenecke, C. Complications after CD19+ CAR T-Cell Therapy. Cancers 2020, 12, 3445.
- Oved, J.H.; Barrett, D.M.; Teachey, D.T. Cellular therapy: Immune-related complications. Immunol. Rev. 2019, 290, 114–126.
- Xu, X.-J.; Tang, Y.-M. Cytokine release syndrome in cancer immunotherapy with chimeric antigen receptor engineered T cells. Cancer Lett. 2014, 343, 172–178.
- Gust, J.; Taraseviciute, A.; Turtle, C.J. Neurotoxicity Associated with CD19-Targeted CAR-T Cell Therapies. CNS Drugs 2018, 32, 1091–1101.
- Gust, J.; Ponce, R.; Liles, W.C.; Garden, G.A.; Turtle, C.J. Cytokines in CAR T Cell–Associated Neurotoxicity. Front. Immunol. 2020, 11, 11.
- Schubert, M.-L.; Schmitt, M.; Wang, L.; Ramos, C.; Jordan, K.; Müller-Tidow, C.; Dreger, P. Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann. Oncol. 2021, 32, 34–48.
- Yu, S.; Yi, M.; Qin, S.; Wu, K. Next generation chimeric antigen receptor T cells: Safety strategies to overcome toxicity. Mol. Cancer 2019, 18, 1–13.
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1- and CTLA-4-Based Inhibitory Chimeric Antigen Receptors (iCARs) Divert Off-Target Immunotherapy Responses. Sci. Transl. Med. 2013, 5, 215ra172.
- Tao, L.; Farooq, M.A.; Gao, Y.X.; Zhang, L.; Niu, C.Y.; Ajmal, I.; Zhou, Y.; He, C.; Zhao, G.X.; Yao, J.; et al. CD19-CAR-T Cells Bearing a KIR/PD-1-Based Inhibitory CAR Eradicate CD19(+)HLA-C1(-) Malignant B Cells While Sparing CD19(+)HLA-C1(+) Healthy B Cells. Cancers 2020, 12, 2612.
- Morsut, L.; Roybal, K.T.; Xiong, X.; Gordley, R.M.; Coyle, S.M.; Thomson, M.; Lim, W.A. Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 2016, 164, 780–791.
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779.
- Srivastava, S.; Salter, A.I.; Liggitt, D.; Yechan-Gunja, S.; Sarvothama, M.; Cooper, K.; Smythe, K.S.; Dudakov, J.A.; Pierce, R.H.; Rader, C.; et al. Logic-Gated ROR1 Chimeric Antigen Receptor Expression Rescues T Cell-Mediated Toxicity to Normal Tissues and Enables Selective Tumor Targeting. Cancer Cell 2019, 35, 489–503.e8.
- Zajc, C.U.; Dobersberger, M.; Schaffner, I.; Mlynek, G.; Puhringer, D.; Salzer, B.; Djinovic-Carugo, K.; Steinberger, P.; Linhares, A.D.; Yang, N.J.; et al. A conformation-specific ON-switch for controlling CAR T cells with an orally available drug. Proc. Natl. Acad. Sci. USA 2020, 117, 14926–14935.
- Giordano-Attianese, G.; Gainza, P.; Gray-Gaillard, E.; Cribioli, E.; Shui, S.; Kim, S.; Kwak, M.-J.; Vollers, S.; Osorio, A.D.J.C.; Reichenbach, P.; et al. A computationally designed chimeric antigen receptor provides a small-molecule safety switch for T-cell therapy. Nat. Biotechnol. 2020, 38, 426–432.
- Sommer, C.; Cheng, H.-Y.; Nguyen, D.; Dettling, D.; Yeung, Y.A.; Sutton, J.; Hamze, M.; Valton, J.; Smith, J.; Djuretic, I.; et al. Allogeneic FLT3 CAR T Cells with an Off-Switch Exhibit Potent Activity against AML and Can Be Depleted to Expedite Bone Marrow Recovery. Mol. Ther. 2020, 28, 2237–2251.
- Gargett, T.; Brown, M.P. The inducible caspase-9 suicide gene system as a "safety switch" to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol. 2014, 5, 235.
- Murty, S.; Labanieh, L.; Murty, T.; Gowrishankar, G.; Haywood, T.; Alam, I.S.; Beinat, C.; Robinson, E.; Aalipour, A.; Klysz, D.D.; et al. PET Reporter Gene Imaging and Ganciclovir-Mediated Ablation of Chimeric Antigen Receptor T Cells in Solid Tumors. Cancer Res. 2020, 80, 4731–4740.
- Richman, S.A.; Wang, L.-C.; Moon, E.K.; Khire, U.R.; Albelda, S.M.; Milone, M.C. Ligand-Induced Degradation of a CAR Permits Reversible Remote Control of CAR T Cell Activity In Vitro and In Vivo. Mol. Ther. 2020, 28, 1600–1613.
- Rodgers, D.T.; Mazagova, M.; Hampton, E.N.; Cao, Y.; Ramadoss, N.S.; Hardy, I.R.; Schulman, A.; Du, J.; Wang, F.; Singer, O.; et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc. Natl. Acad. Sci. USA 2016, 113, E459–E468.
- Mestermann, K.; Giavridis, T.; Weber, J.; Rydzek, J.; Frenz, S.; Nerreter, T.; Mades, A.; Sadelain, M.; Einsele, H.; Hudecek, M. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med. 2019, 11, eaau5907.
- Salzer, B.; Schueller, C.M.; Zajc, C.U.; Peters, T.; Schoeber, M.A.; Kovacic, B.; Buri, M.C.; Lobner, E.; Dushek, O.; Huppa, J.B.; et al. Engineering AvidCARs for combinatorial antigen recognition and reversible control of CAR function. Nat. Commun. 2020, 11, 1–16.
- Feldmann, A.; Hoffmann, A.; Bergmann, R.; Koristka, S.; Berndt, N.; Arndt, C.; Loureiro, L.R.; Kittel-Boselli, E.; Mitwasi, N.; Kegler, A.; et al. Versatile chimeric antigen receptor platform for controllable and combinatorial T cell therapy. OncoImmunology 2020, 9, 1785608.
- Albert, S.; Arndt, C.; Feldmann, A.; Bergmann, R.; Bachmann, D.; Koristka, S.; Ludwig, F.; Ziller-Walter, P.; Kegler, A.; Gärtner, S.; et al. A novel nanobody-based target module for retargeting of T lymphocytes to EGFR-expressing cancer cells via the modular UniCAR platform. OncoImmunology 2017, 6, e1287246.
- Kim, M.S.; Ma, J.S.Y.; Yun, H.; Cao, Y.; Kim, J.Y.; Chi, V.; Wang, D.; Woods, A.; Sherwood, L.; Caballero, D.; et al. Redirection of Genetically Engineered CAR-T Cells Using Bifunctional Small Molecules. J. Am. Chem. Soc. 2015, 137, 2832–2835.
- Ma, J.S.Y.; Kim, J.Y.; Kazane, S.A.; Choi, S.-H.; Yun, H.Y.; Kim, M.S.; Rodgers, D.T.; Pugh, H.M.; Singer, O.; Sun, S.B.; et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E450–E458.
- Roddie, C.; O’Reilly, M.; Pinto, J.D.A.; Vispute, K.; Lowdell, M. Manufacturing chimeric antigen receptor T cells: Issues and challenges. Cytotherapy 2019, 21, 327–340.
- Kacherovsky, N.; Cardle, I.I.; Cheng, E.L.; Yu, J.L.; Baldwin, M.L.; Salipante, S.J.; Jensen, M.C.; Pun, S.H. Traceless aptamer-mediated isolation of CD8+ T cells for chimeric antigen receptor T-cell therapy. Nat. Biomed. Eng. 2019, 3, 783–795.
- Zhao, J.; Lin, Q.; Song, Y.; Liu, D. Universal CARs, universal T cells, and universal CAR T cells. J. Hematol. Oncol. 2018, 11, 1–9.
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199.
- From Pluripotent Stem to CAR T Cells. Cancer Discov. 2018, 8, OF5.
- Feucht, J.; Sun, J.; Eyquem, J.; Ho, Y.-J.; Zhao, Z.; Leibold, J.; Dobrin, A.; Cabriolu, A.; Hamieh, M.; Sadelain, M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019, 25, 82–88.
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; Van Der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, K.M.C.M.; Sadelain, J.E.J.M.-S.T.G.S.J.C.V.D.S.M.H.A.O.M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nat. Cell Biol. 2017, 543, 113–117.
- Stenger, D.; Stief, T.A.; Kaeuferle, T.; Willier, S.; Rataj, F.; Schober, K.; Vick, B.; Lotfi, R.; Wagner, B.; Grünewald, T.G.P.; et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood 2020, 136, 1407–1418.
- Chang, C.; Van Der Stegen, S.; Mili, M.; Clarke, R.; Lai, Y.-S.; Witty, A.; Lindenbergh, P.; Yang, B.-H.; Husain, M.; Shaked, H.; et al. FT819: Translation of Off-the-Shelf TCR-Less Trac-1XX CAR-T Cells in Support of First-of-Kind Phase I Clinical Trial. Blood 2019, 134, 4434.
- Ferreira, M.V.; Cabral, E.T.; Coroadinha, A.S. Progress and Perspectives in the Development of Lentiviral Vector Producer Cells. Biotechnol. J. 2021, 16.
- Harris, E.; Elmer, J.J. Optimization of electroporation and other non-viral gene delivery strategies for T cells. Biotechnol. Prog. 2021, 37, 37.
- Bishop, D.C.; Caproni, L.; Gowrishankar, K.; Legiewicz, M.; Karbowniczek, K.; Tite, J.; Gottlieb, D.J.; Micklethwaite, K.P. CAR T Cell Generation by piggyBac Transposition from Linear Doggybone DNA Vectors Requires Transposon DNA-Flanking Regions. Mol. Ther. Methods Clin. Dev. 2020, 17, 359–368.
- Querques, I.; Mades, A.; Zuliani, C.; Miskey, C.; Alb, M.; Grueso, E.; Machwirth, M.; Rausch, T.; Einsele, H.; Ivics, Z.; et al. A highly soluble Sleeping Beauty transposase improves control of gene insertion. Nat. Biotechnol. 2019, 37, 1502–1512.
- Zhang, J.; Hu, Y.; Yang, J.; Li, W.; Tian, Y.; Wei, G.; Zhang, L.; Zhao, K.; Qi, Y.; Tan, B.; et al. Development and clinical evaluation of non-viral genome specific targeted CAR T cells in relapsed/refractory B-cell non-Hodgkin lymphoma. medRxiv 2020.
- Dai, X.; Park, J.J.; Du, Y.; Kim, H.R.; Wang, G.; Errami, Y.; Chen, S. One-step generation of modular CAR-T cells with AAV–Cpf1. Nat. Methods 2019, 16, 247–254.
- Moffett, H.F.; Coon, M.E.; Radtke, S.; Stephan, S.B.; McKnight, L.; Lambert, A.; Stoddard, B.L.; Kiem, H.P.; Stephan, M.T. Hit-and-run programming of therapeutic cytoreagents using mRNA nanocarriers. Nat. Commun. 2017, 8, 1–13.

