 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mahmoud Alhosin | + 3140 word(s) | 3140 | 2021-04-22 04:38:07 | | | |
| 2 | Nora Tang | Meta information modification | 3140 | 2021-05-27 05:26:13 | | |
Video Upload Options
The tumor suppressor TAp73 is a member of the p53 family, which is inhibited in many human solid and hematological tumors. In contrast to those in the p53 gene, mutations in the p73 gene are very rare in tumors, suggesting that the decrease in TAp73 activity and expression detected in those tumors are caused mainly by coordinated post-translational modifications of TAp73.
1. Introduction
The p53 gene, commonly referred to as the guardian of the human genome, is highly mutated in approximately 50% of human cancers [1][2]. It is a vital tumor-suppressor gene due to the ability of p53 to induce apoptosis and growth arrest in response to cell stress, genotoxicity, and DNA damage [3]. Overall, p53 is a master transcription factor of cell cycle inhibitors, such as p21WAF1 [4][5], as well as several pro-apoptotic genes, including Bax and Bim [6][7]. p53 was thought to be a unique gene in the human genome for more than two decades until the discovery of two additional members of its family, p63 and p73, which share a similar structure and function [8][9][10][11]. The tumor suppressor p53 and its homologs, p63 and p73, play crucial roles in the regulation of several cellular activities such as DNA damage response, development, cellular homeostasis, aging, and metabolism [12][13]. Like other members, p73 is also expressed as multiple functional isoforms (Figure 1). The p73 gene has two different promoters, thereby allowing for the generation of two types of isoforms with two opposite functions. The transactivation-p73 (TAp73) isoforms contain the N-terminal transactivation domain with pro-apoptotic functions, whereas the N-terminally truncated p73 (ΔNp73) isoforms have anti-apoptotic properties (Figure 1) [11][14][15][16][17][18].
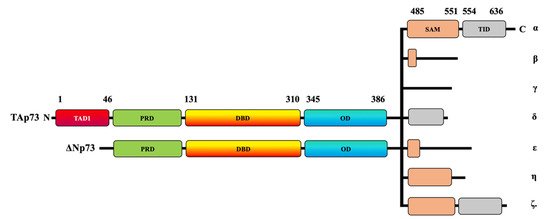
Figure 1. Schematic representation of TAp73 and ΔNp73 domains. The human p73 protein comprises transcriptional activating domains (TADs) and a proline-rich domain (PRD) at its N-terminal and a DNA-binding domain (DBD). The p73 C-terminal domain is composed of an oligomerization domain (OD), a sterile alpha motif (SAM) domain, and a transactivation inhibitory domain (TID). At least two N-terminal isoforms, TAp73 and ΔNp73, can be combined with the C-terminal α, β, γ, δ, ε, ζ, and η isoforms.
The human p73 gene contains 14 exons that extend to around 80 kb. The p73 protein contains approximately eight domains. At its N-terminal, the human p73 protein comprises transcriptional activating domains (TADs), a proline-rich domain (PRD), and a DNA-binding domain (DBD). The carboxy-terminal (C-terminal) is composed of an oligomerization domain (OD), a sterile alpha motif (SAM) domain, and a transactivation-inhibitory domain (TID) [19][20]. The TID is a post-SAM region and is absent in p53 [20]. The TAp73 isoforms have multiple variants—α, β, γ, δ, ε, ζ, and η—that are expressed due to alternative splicing on the SAM domain located at the C-terminal region [21] (Figure 1). TAp73α is considered the longest variant because it has the longest SAM domain and the longest C-terminal, whereas p73β has a shorter SAM domain and a shorter C-terminal tail [20][22].
ΔNp73 lacks a transactivation domain (Figure 1), but it has oncogenic potential, as displayed by its dominant-negative effects on both wildtype p53 and TAp73 [16][17]. Like TAp73, ΔNp73 can be combined with C-terminal isoforms (Figure 1). ΔNp73 usually displaces wildtype p53 and TAp73 at the oligomerization level through competition for the DNA-binding site [23]. The overexpression of p73 induces apoptosis, whereas ΔNp73 can inhibit p73-induced cell death; therefore, the fine balance between the two variants can determine cell fate.
The increasing interest in the p73 gene has resulted from the very low frequency of its mutations in human cancers [21][24][25]. Indeed, mutations in the p73 gene are very rare in human tumors compared to those in the p53 gene, which have been detected in more than 50% of human cancers [2][26][27]. In addition to p73 gene mutations, p73 has been also found to be epigenetically silenced in some leukemias and lymphomas through the hypermethylation of its promoter [28][29][30]. Alternatively, the loss of p73 in cancer may result from several post-translational modifications, including the ubiquitin-dependent proteasomal degradation pathway, phosphorylation, acetylation, and small ubiquitin-related modifier (SUMO)ylation. Thus, a deep understanding of p73 post-translational modifications will be extremely helpful in finding and developing new strategies for the prevention and treatment of cancers, especially those with p53 mutations. In this review, we summarize the current knowledge regarding the different pathways implicated in the regulation of p73 at the post-translational level. This review also highlights the growing importance of targeting the post-translational modifications of p73 as a promising antitumor strategy, regardless of the p53 status.
2. Ubiquitination-Dependent p73 Inhibition
In all cells, the intracellular proteins are degraded through the ubiquitin-proteasome pathway (UPP). The UPP contains specific enzymes that bind polypeptide chains to ubiquitin (Ub) and tag them for proteasomal degradation [31][32]. The tagged polypeptides are recognized by the 26S proteasome, a multicatalytic protease complex that degrades large ubiquitinated polypeptides into small peptides. Three enzymes are required for the ubiquitin tagging process: E1, a Ub-activating enzyme; E2, a Ub carrier or conjugating protein; and E3, a Ub protein ligase [33]. The E3 ligases are responsible for ligating Ub to polypeptides or proteins. By contrast, the deubiquitinating enzymes (DUBs) remove Ub from ubiquitinated proteins, thereby promoting protein stability.
The reversible regulation of Ub by E3 ligases and DUBs has received increasing attention for several diseases, including cancers [34][35][36][37]. The ubiquitination pathway affects many tumor-suppressor proteins, including p53 and its homologs, p63 and p73 [38]. Over the last 20 years, researchers have discovered many ubiquitin E3 ligases that directly promote the protein degradation of p53, p63, and p73 by either ubiquitination-dependent or ubiquitination-independent proteasome pathways [39]. TAp73 was shown to undergo ubiquitination and to be inhibited by various E3 ubiquitin-ligase enzymes (Table 1) (Figure 2), such as Itch [40][41][42][43], mouse double minute 2 homolog (MDM2) and its homologue mouse double minute X (MDMX) [44][45][46], p53-induced RING-H2 protein (Pirh2) [47][48][49], tripartite motif 32 (TRIM32) [50], FBXO protein 45 (FBXO45) [51], Cullin4A (cul4A)-dependent ligase (CDL4A) [52], and WW Domain Containing E3 Ubiquitin Protein Ligase 2(WWP2) [53]. PIR2 (p73-induced ring protein 2) is also an E3 ubiquitin ligase for p73 through a mechanism that involves targeting the ΔNp73/TAp73α balance rather than the direct ubiquitination of p73 [54].
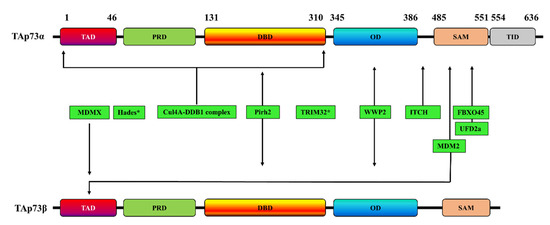
Figure 2. Schematic representation of the interaction sites of various E3 ubiquitin-ligases with TAp73α, TAp73β, or both. * indicates that the p73 interaction site is unknown. TAD: transactivation domain; PRD: proline rich domain; DBD: DNA binding domain; OD: oligomerization domain; SAM: sterile alpha motif; TID: transactivation inhibitory domain.
Table 1. Role of E3 ligases in p73 stability. TRIM32: tripartite motif 32; Cul4A: Cullin4A; MDM2: mouse double minute 2 homolog; Pirh2: p53-induced RING-H2 protein.
| E3 Ligase | TAp73 Isoform | p73 Interaction Site | Effects on p73 | Refs |
|---|---|---|---|---|
| ITCH | p73α | PY motif (Met 452–Ala 489) | Proteasome-dependent p73 degradation | [43] |
| MDM2 | p73α | - | p73α activity inhibition but without promoting its degradation | [55][56] |
| p73β | Two residues (F15 and W19) | p73β activity inhibition but without promoting its degradation | [57] | |
| p73α | - | p73 degradation through a mechanism involving the interaction of MDM2 with Itch | [45] | |
| p73α and p73β | N-terminal domain | p73 ubiquitination but without promoting its degradation | [44] | |
| p73α | The C-terminal SAM domain (especially the peptide comprising α4 and α5 helices of SAM) | - | [58] | |
| MDMX | p73α | TA domain | Subcellular localization of p73 and inactivation of transcription | [46][59] |
| Pirh2 | p73α and p73β | DNA-binding domain | p73 ubiquitination and repressing p73-dependent transcriptional activity | [47] |
| TRIM32 | p73α | - | p73 ubiquitination and degradation, inhibiting p73 transcriptional activity | [50] |
| FBXO45 | p73α | SAM domain | Proteasome-dependent degradation of p73 | [51] |
| UFD2a | p73α | SAM domain | Ubiquitination-independent p73 degradation | [60] |
| Hades | - | - | Ubiquitination of p73, promoting its degradation | [61] |
| Cul4A–DDB1 complex | p73α | The N-terminal region of p73, encompassing residues 1–320 and including the DNA-binding domain. | p73 monoubiquitination and inhibition of its transcriptional activity without affecting p73 stability | [52] |
| WWP2 | p73α and p73β | Oligomerization domain | p73 ubiquitination and its proteasomal degradation in | [53] |
2.1. Itch
Itch E3 ubiquitin-protein ligase (Itch) has been reported to be highly expressed in several tumors, including lung cancer, breast cancer, pancreatic cancer, skin cancer, and neuroblastoma, and its inhibition leads to the induction of apoptosis and the inhibition of cell proliferation [38][59][62][63][64]. Itch selectively binds and ubiquitinates p73α via the PY motif (Met 452–Ala 489) but not p53, thereby leading to a proteasome-dependent p73 degradation [43]. The use of the proteasome inhibitor MG132 or the depletion of Itch using siRNA causes the inhibition of the Itch-mediated p73 degradation and increases p73 expression levels [43]. Similarly, treating cancer cells with DNA-damaging agents, such as doxorubicin, cisplatin, and etoposide, decreases the expression of endogenous Itch in a time- and dose-dependent manner, and this effect is associated with an increase in endogenous p73 levels and enhanced apoptosis [43].
Itch and p73 expressions are controlled through several mechanisms. For example, the Itch–p73 interaction was proposed to be under the control of the transcriptional co-activator Yes-associated protein 1 (Yap1), another partner of p73 [40]. Indeed, Yap1 competes with Itch for binding to p73, thereby preventing the Itch-mediated ubiquitination and degradation of p73. Cisplatin increases p73 expression and apoptosis, while the depletion of Yap1 using siRNA blocks these effects [40]. Yap1 interacts with another transcription activator, Runx, to target the promoter of Itch and enhance its transcription, with subsequent support of the degradation of p73 [41].Nedd4-binding partner-1 (N4BP1) has been also found to compete with Itch for binding to p73α, thus reducing the ability of Itch to interact with and ubiquitylate p73α [65]. The Itch/p73 axis is also regulated in chronic lymphocytic leukemia (CLL) cells through the direct regulation of Itch by microRNA 106b (miR106b) [42]. Indeed, the induction of miR106b in CLL cells is associated with a decrease in Itch expression and an increase in the expression of p73 and PUMA, as well as induction of CLL apoptosis [42].
2.2. MDM2/MDMX
MDM2 is a negative regulator of p53; its interaction with the TAD of p53 leads to a ubiquitination-mediated proteasomal degradation of p53 [13][66]. MDM2 also interacts with p73α and inhibits its function, but it does so without promoting p73 degradation [55][56]. MDM2 also forms a specific complex in vitro and in vivo with p73β through at least two residues (F15 and W19) [57]. MDM2 overexpression reduces the ability of p73β to activate transcription but without promoting its degradation, indicating that MDM2 can directly induce the degradation of the p53 protein but not of its homolog p73 [57]. However, MDM2 has been shown to induce p73 degradation in HeLa cells through a mechanism involving the interaction of MDM2 with Itch [45].
Adriamycin can increase p73 expression and HeLa cell apoptosis, most likely by decreasing the expression of both MDM2 and Itch. Similarly, the MDM2-mediated p73 degradation is inhibited in cells treated with the inhibitor proteasome MG132 [45]. In the same context, MDM2 was reported to use its RING domain to bind to p73α and p73β at multiple Lys residues of ubiquitin, thereby promoting the ubiquitination of both isoforms but without inducing their degradation [44]. MDM2 overexpression induces Itch-mediated p73 degradation in MDM2-deficient mouse embryo fibroblasts, whereas its depletion using siRNA induces p73-dependent transactivation and apoptosis [44].
A recent study showed that the MDM2/p73 interaction is established by specific binding between the N-terminus TA domain of MDM2 and the SAM domain of p73 [58]. MDMX, the structural homolog of MDM2, was also shown to negatively regulate p73 [46][59][67]. MDMX physically interacts with p73α, and its overexpression specifically induces alterations in the subcellular localization of p73α but not of the p73 homologs p53 and p63 [59]. Both MDM2 and MDMX are capable of binding to the TA domain of p73 [46].
2.3. Pirh2
Pirh2 is a RING finger E3 ligase that induces p53 degradation by promoting its ubiquitination [68][69]. Several studies have reported that Pirh2 acts as an oncogene, and its overexpression in cancer is associated with decreased p53 expression levels [48][49][70]. Pirh2 has also been shown to interact with the DNA-binding domain of both p73α and p73β [68][71][72], and its E3 ligase activity promotes p73 ubiquitination both in vivo and in vitro [47][73]. Pirh2 ubiquitinates p73 in vitro via K63-linked chains, while it promotes p73 ubiquitination in vivo by targeting K11-, K29-, K48-, and K63-linked chains [47]. The downregulation of Pirh2 significantly decreases p73 ubiquitination, whereas its overexpression is correlated with an inhibition of p73-dependent translational activity [47]. Pirh2 knockdown in the RKO colon carcinoma cell line inhibits cell growth and increases the levels of p73 and the cell cycle inhibitor p21WAF1 [73]. Conversely, the overexpression of Pirh2 in colon and breast cells induces the degradation of p73 via the proteasomal pathway [73]. The exposure of RKO cells to either doxorubicin or camptothecin decreases Pirh2-mediated p73 polyubiquitination [73].
2.4. TRIM32
Tripartite Motif Containing 32 (TRIM32) a RING protein, is overexpressed in several tumors, including skin tumors. It has E3 ubiquitin ligase proprieties and plays a role in tumor growth, metastasis, and resistance to anti-cancer drugs [74]. In this context, TRIM32 is involved in the ubiquitination of the tumor-suppressor Abl-interactor 2 (Abi2), resulting in an enhancement of cell proliferation and metastases [75]. TRIM32 also regulates the expression of p73 through the ubiquitination pathway, thereby promoting cell survival and tumor growth [50][76]. A regulatory feedback loop has been reported between TP73 and TRIM32 in human embryonic kidney 293 cells [50]. In HEK293 cells, p73 can act as a transcriptional regulator of TRIM32, and the depletion of p73 significantly decreases TRIM32 expression. TRIM32, in turn, binds to p73α and promotes its ubiquitination and degradation, thus inhibiting p73 transcriptional activity [50]. The overexpression of TRIM32 inhibits the p73 transcriptional activation of the p21WAF1 promoter, thereby enhancing cell cycle progression and cell survival [50].
2.5. FBXO45
Several studies have reported the overexpression of FBXO45 in human cancer and an essential role for FBXO45 in tumorigenesis and progression [77][78][79]. FBXO45 induces the ubiquitination–proteasomal degradation of several tumor-suppressor proteins, such as Par4 (prostate apoptosis response protein 4) [80], F-box/WD repeat-containing protein 7 (FBXW7) [81], and p73 [51]. Through its spla and the ryanodine receptor (SPRY) domain, FBXO45 specifically binds to the SAM domain of p73α, but not to its homolog p53, thus leading to the proteasome-dependent degradation of p73 both in vitro and in vivo [51]. The silencing of FBXO45 in the BT-20 cells, a p53-mutated cell line, induces the expression of p73 at the protein level [51]. The exposure of FBO45-depleted BT-20 cells to doxorubicin induces apoptosis, indicating that FBXO45 specifically binds to p73 and induces apoptosis via a p73-dependent mechanism [51]. Similar to p73, FBXO45 is also able to interact with ΔNp73 to play a crucial role in its stability in apoptosis and the DNA damage response. However, mutant FBXO45 has shown an impaired function against p73 and does not cause ubiquitination-associated p73 degradation [51].
2.6. UFD2a
Ubiquitin factor 2A (UFD2a), also known as Ubiquitin factor E4B (UBE4B), is an E3/E4 ligase belonging to the U box-type ubiquitin protein ligase family that can catalyze the elongation of the polyubiquitin chain. In response to apoptosis, UFD2a is cleaved by caspase 6, resulting in impaired enzymatic activity and dysfunction, as well as confirming the role of UFD2a in apoptotic signaling [82]. UFD2a physically interacts with p53 in brain tumors to promote p53 polyubiquitination and degradation, thus decreasing p53-mediated apoptotic activity [83]. Similar to its effects on p53, UFD2a regulates the stability of p73 via ubiquitination-independent degradation [60]. UFD2a also physically interacts with the SAM domain of p73α [60]. The treatment of SH-SY5Y neuroblastoma cells with cisplatin results in a decrease in the expression of UFD2a, while p73 protein expression and apoptosis are increased [60]. The expression of the p73 protein is also increased in UFD2a-depleted cells [60]. Alternatively, the overexpression of UFD2a decreases both p73α expression and p73-mediated pro-apoptotic activity [60]. Similar findings have been reported in squamous cell carcinoma (SCC) cells [84]. UFD2a knockdown in SCC cells was found to result in p73 accumulation and sensitized the SCC cells to the inhibitory effects of 1,25D3 (the most active metabolite of vitamin D) on cell growth, suggesting that the UFD2a/p73 pathway contributes to the antiproliferative effects of anticancer drugs [84][85].
2.7. Hades
Hades, also referred to as mitochondrial ubiquitin ligase activator of NFKB1 (MULAN), is an E3 ubiquitin ligase. Hades is anchored to the outer membrane of mitochondria through its transmembrane domains, with a conserved C-terminal RING-finger domain with an E3 ligase activity facing the cytosol [61]. Hades interacts with p53 in the mitochondria to reduce p53 stability [86]. Hades polyubiquitinates p53 through an MDM2-independent mechanism, thereby inhibiting the p53-dependent mitochondrial cell death pathway by blocking the interaction between p53 and Bcl-2 [86]. Through its RING-finger domain, Hades also interacts with p73 in etoposide-treated H1299 cells [61]. In response to etoposide treatment, p73 is translocated to the mitochondria to colocalize with Hades [61]. Hades induces the ubiquitination of p73 and its degradation, whereas its depletion using siRNA significantly inhibits p73 degradation, indicating that Hades is involved in the ubiquitination-dependent degradation of p73 [61].
2.8. The Cul4A–DDB1 E3 Ubiquitin Ligase Complex
The CDL4A complex targets various proteins that are involved in several signaling pathways, including the DNA damage response and the cell cycle [87][88][89], and it contributes to the degradation of several proteins through the ubiquitin-proteasome pathway. In this context, Cul4A has been reported to interact with p53 to promote its proteasomal degradation [90]. Through damage specific DNA binding protein 1(DDB1), the CDL4A complex can directly bind to the N-terminal region of p73, which encompasses residues 1–320 and includes the DNA-binding domain, thereby inducing p73 mono-ubiquitination and inhibiting p73-dependent transcriptional activity without affecting p73 stability at the protein level [52]. The depletion of DDB1 induces the expression of several known target genes of p73, such as p21WAF1 and PUMA [52]. Clinically, high expression levels of Cul4A in human breast carcinomas are associated with the repression of target genes of p73, indicating that Cul4A is a potent inhibitor of p73 transcriptional activity in cancer [52].
2.9. WWP2
WWP2, an E3 ubiquitin ligase, has an important role in different cellular functions and ubiquitinates, and it decreases the expression of the tumor-suppressor genes PTEN [91] and p73 [53] through the proteasomal pathway. Through its WW3 domain, WWP2 specifically interacts with the oligomerization domain of both p73α and p73β but not with p53 [51]. This results in p73 ubiquitination via a K48 linkage, with the subsequent induction of p73 degradation in the proteasome [53]. Interestingly, WWP2 depletion dramatically decreases p73 ubiquitination and increases its expression at the protein level [53].
3. Conclusions
The downregulation of the tumor suppressor TAp73 represses the transcription of several anti-proliferative and pro-apoptotic genes in many human solid and hematological tumors. This, in turn, inhibits apoptosis and enhances cell proliferation. Via its structural domains, which show a high degree of similarity with those of p53, TAp73 interacts with several E3 ubiquitin ligases, kinases, and deacetylases. These proteins, which work together to orchestrate the multiple anti-proliferative and pro-apoptotic functions of TAp73, also offer multiple promising candidates for cancer therapy. Since mutations in the p73 gene are very rare in tumors, unlike the case for the p53 gene, the decrease in both activity and quantity of TAp73 detected in various human tumors could arise, in large part, from coordinated post-translational modifications of TAp73. This review has highlighted the multiple post-translational modifications underlying TAp73 regulation in cancer cells and the growing importance of targeting their trigger enzymes as a promising antitumor strategy. Thus, understanding the post-translational modifications involved in TAp73 regulation will allow for the identification of new targets and the exploration of new drugs that can increase the expression of TAp73. This will allow cancer cells to undergo apoptosis, regardless of their p53 status, through the reactivation of several pro-apoptotic genes.
References
- Olivier, M.; Hussain, S.P.; De Fromentel, C.C.; Hainaut, P.; Harris, C.C. TP53 mutation spectra and load: A tool for generating hypotheses on the etiology of cancer. IARC Sci. Publ. 2004, 2004, 247–270.
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harb. Perspect. Biol. 2009, 2, a001008.
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. Dissecting p53 tumor suppressor functions in vivo. Cancer Cell 2002, 1, 289–298.
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825.
- Harper, J.W.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge, S.J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cy-clin-dependent kinases. Cell 1993, 75, 805–816.
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct Activation of Bax by p53 Mediates Mitochondrial Membrane Permeabilization and Apoptosis. Science 2004, 303, 1010–1014.
- Han, J.; Goldstein, L.A.; Hou, W.; Gastman, B.R.; Rabinowich, H. Regulation of Mitochondrial Apoptotic Events by p53-mediated Disruption of Complexes between Antiapoptotic Bcl-2 Members and Bim. J. Biol. Chem. 2010, 285, 22473–22483.
- Kelley, M.L.; Winge, P.; Heaney, J.D.; Stephens, R.E.; Farell, J.H.; Van Beneden, R.J.; Reinisch, C.L.; Lesser, M.P.; Walker, C.W. Expression of homologues for p53 and p73 in the softshell clam (Mya arenaria), a naturally-occurring model for human cancer. Oncogene 2001, 20, 748–758.
- Kaghad, M.; Bonnet, H.; Yang, A.; Creancier, L.; Biscan, J.-C.; Valent, A.; Minty, A.; Chalon, P.; Lelias, J.-M.; Dumont, X.; et al. Monoallelically Expressed Gene Related to p53 at 1p36, a Region Frequently Deleted in Neuroblastoma and Other Human Cancers. Cell 1997, 90, 809–819.
- Schmale, H.; Bamberger, C. A novel protein with strong homology to the tumor suppressor p53. Oncogene 1997, 15, 1363–1367.
- Yang, A.; Kaghad, M.; Wang, Y.; Gillett, E.; Fleming, M.D.; Dötsch, V.; Andrews, N.C.; Caput, D.; McKeon, F. p63, a p53 Homolog at 3q27–29, Encodes Multiple Products with Transactivating, Death-Inducing, and Dominant-Negative Activities. Mol. Cell 1998, 2, 305–316.
- Lunghi, P.; Costanzo, A.; Mazzera, L.; Rizzoli, V.; Levrero, M.; Bonati, A. The p53 Family Protein p73 Provides New Insights into Cancer Chemosensitivity and Targeting. Clin. Cancer Res. 2009, 15, 6495–6502.
- De Laurenzi, V.; Melino, G. Evolution of functions within the p53/p63/p73 family. Ann. N. Y. Acad. Sci. 2000, 926, 90–100.
- Ishimoto, O.; Kawahara, C.; Enjo, K.; Obinata, M.; Nukiwa, T.; Ikawa, S. Possible oncogenic potential of DeltaNp73: A newly identified isoform of human p73. Cancer Res. 2002, 62, 636–641.
- Moll, U.M.; Slade, N. p63 and p73: Roles in development and tumor formation. Mol. Cancer Res. 2004, 2, 371–386.
- Grob, T.J.; Novak, U.; Maisse, C.; Barcaroli, D.; Lüthi, A.U.; Pirnia, F.; Hügli, B.; Graber, H.U.; De Laurenzi, V.; Fey, M.F.; et al. Human delta Np73 regulates a dominant negative feedback loop for TAp73 and p53. Cell Death Differ. 2001, 8, 1213–1223.
- Zaika, A.I.; Slade, N.; Erster, S.H.; Sansome, C.; Joseph, T.W.; Pearl, M.; Chalas, E.; Moll, U.M. ΔNp73, A Dominant-Negative Inhibitor of Wild-type p53 and TAp73, Is Up-regulated in Human Tumors. J. Exp. Med. 2002, 196, 765–780.
- Ramos, H.; Raimundo, L.; Saraiva, L. p73: From the p53 shadow to a major pharmacological target in anticancer therapy. Pharmacol. Res. 2020, 162, 105245.
- DeYoung, M.P.; Ellisen, L.W. p63 and p73 in human cancer: Defining the network. Oncogene 2007, 26, 5169–5183.
- Yoon, M.-K.; Ha, J.-H.; Lee, M.-S.; Chi, S.-W. Structure and apoptotic function of p73. BMB Rep. 2015, 48, 81–90.
- Conforti, F.; Yang, A.L.; Agostini, M.; Rufini, A.; Tucci, P.; Nicklison-Chirou, M.V.; Grespi, F.; Velletri, T.; Knight, R.A.; Melino, G.; et al. Relative expression of TAp73 and ΔNp73 isoforms. Aging 2012, 4, 202–205.
- Ozaki, T.; Nakagawara, A. p73, a sophisticated p53 family member in the cancer world. Cancer Sci. 2005, 96, 729–737.
- Müller, M.; Schleithoff, E.S.; Stremmel, W.; Melino, G.; Krammer, P.H.; Schilling, T. One, two, three-p53, p63, p73 and chemosensi-tivity. Drug Resist Updates 2006, 9, 288–306.
- Davis, P.K.; Dowdy, S.F. p73. Int. J. Biochem. Cell Biol. 2001, 33, 935–939.
- Coates, P.; Coates, P. Regulating p73 isoforms in human tumours. J. Pathol. 2006, 210, 385–389.
- Greenblatt, M.S.; Bennett, W.P.; Hollstein, M.; Harris, C.C. Mutations in the p53 tumor suppressor gene: Clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994, 54, 4855–4878.
- Pitolli, C.; Wang, Y.; Mancini, M.; Shi, Y.; Melino, G.; Amelio, I. Do Mutations Turn p53 into an Oncogene? Int. J. Mol. Sci. 2019, 20, 6241.
- Kawano, S.; Miller, C.W.; Gombart, A.F.; Bartram, C.R.; Matsuo, Y.; Asou, H.; Sakashita, A.; Said, J.; Tatsumi, E.; Koeffler, H.P. Loss of p73 gene expression in leukemias/lymphomas due to hypermethylation. Blood 1999, 94, 1113–1120.
- Pluta, A.; Nyman, U.; Joseph, B.; Robak, T.; Zhivotovsky, B.; Smolewski, P. The role of p73 in hematological malignancies. Leukemia 2006, 20, 757–766.
- Liu, M.; Taketani, T.; Li, R.; Takita, J.; Taki, T.; Yang, H.W.; Kawaguchi, H.; Ida, K.; Matsuo, Y.; Hayashi, Y. Loss of p73 gene expression in lymphoid leukemia cell lines is associated with hypermethylation. Leuk. Res. 2001, 25, 441–447.
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein Degradation by the Ubiquitin–Proteasome Pathway in Normal and Disease States. J. Am. Soc. Nephrol. 2006, 17, 1807–1819.
- Masclef, L.; Maxime, U.; Ahmed, O.; Nkwe, N.S.; Barbour, H.; Iannantuono, N.V.G.; Boubekeur, A.; Daou, S.; Affar, E.B. In vitro ubiq-uitination and deubiquitination assays of nucleosomal histones. J. Vis. Exp. 2019, 2019.
- Park, H.-B.; Kim, J.-W.; Baek, K.-H. Regulation of Wnt Signaling through Ubiquitination and Deubiquitination in Cancers. Int. J. Mol. Sci. 2020, 21, 3904.
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229.
- Suresh, B.; Lee, J.; Kim, K.S.; Ramakrishna, S. The Importance of Ubiquitination and Deubiquitination in Cellular Reprogram-ming. Stem Cells Int. 2016, 2016, 6705927.
- Mennerich, D.; Kubaichuk, K.; Kietzmann, T. DUBs, Hypoxia, and Cancer. Trends Cancer 2019, 5, 632–653.
- Kubaichuk, K.; Kietzmann, T. Involvement of E3 Ligases and Deubiquitinases in the Control of HIF-α Subunit Abundance. Cells 2019, 8, 598.
- Oberst, A.; Rossi, M.; Salomoni, P.; Pandolfi, P.P.; Oren, M.; Melino, G.; Bernassola, F. Regulation of the p73 protein stability and degradation. Biochem. Biophys. Res. Commun. 2005, 331, 707–712.
- Antoniou, N.; Lagopati, N.; Balourdas, D.I.; Nikolaou, M.; Papalampros, A.; Vasileiou, P.V.S.; Myrianthopoulos, V.; Kotsinas, A.; Shiloh, Y.; Liontos, M.; et al. The Role of E3, E4 Ubiquitin Ligase (UBE4B) in Human Pathologies. Cancers 2019, 12, 62.
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. The Yes-associated protein 1 stabilizes p73 by preventing Itch-mediated ubiqui-tination of p73. Cell Death Differ. 2007, 14, 743–751.
- Levy, D.; Reuven, N.; Shaul, Y. A Regulatory Circuit Controlling Itch-mediated p73 Degradation by Runx. J. Biol. Chem. 2008, 283, 27462–27468.
- Sampath, D.; Calin, G.A.; Puduvalli, V.K.; Gopisetty, G.; Taccioli, C.; Liu, C.G.; Ewald, B.; Liu, C.; Keating, M.J.; Plunkett, W. Specific acti-vation of microRNA106b enables the p73 apoptotic response in chronic lymphocytic leukemia by targeting the ubiquitin ligase Itch for degradation. Blood 2009, 113, 3744–3753.
- Rossi, M.; De Laurenzi, V.; Munarriz, E.; Green, D.R.; Liu, Y.-C.; Vousden, K.H.; Cesareni, G.; Melino, G. The ubiquitin–protein ligase Itch regulates p73 stability. EMBO J. 2005, 24, 836–848.
- Wu, H.; Leng, R.P. MDM2 mediates p73 ubiquitination: A new molecular mechanism for suppression of p73 function. Oncotarget 2015, 6, 21479–21492.
- Kubo, N.; Okoshi, R.; Nakashima, K.; Shimozato, O.; Nakagawara, A.; Ozaki, T. MDM2 promotes the proteasomal degradation of p73 through the interaction with Itch in HeLa cells. Biochem. Biophys. Res. Commun. 2010, 403, 405–411.
- Zdzalik, M.; Pustelny, K.; Kedracka-Krok, S.; Huben, K.; Pecak, A.; Wladyka, B.; Jankowski, S.; Dubin, A.; Potempa, J.; Dubin, G. Inter-action of regulators Mdm2 and Mdmx with transcription factors p53, p63 and p73. Cell Cycle 2010, 9, 4584–4591.
- Wu, H.; Abou, Z.R.; Flores, E.R.; Leng, R.P. Pirh2, a Ubiquitin E3 Ligase, Inhibits p73 Transcriptional Activity by Promoting Its Ubiquitination. Mol. Cancer Res. 2011, 9, 1780–1790.
- Jung, Y.-S.; Qian, Y.; Chen, X. Pirh2 RING-finger E3 ubiquitin ligase: Its role in tumorigenesis and cancer therapy. FEBS Lett. 2012, 586, 1397–1402.
- Halaby, M.J.; Hakem, R.; Hakem, A. Pirh2: An E3 ligase with central roles in the regulation of cell cycle, DNA damage response, and differentiation. Cell Cycle 2013, 12, 2733–2737.
- Gonzalez-Cano, L.; Hillje, A.-L.; Álvarez, S.F.; Marques, M.M.; Blanch, A.; Ian, R.W.; Irwin, M.S.; Schwamborn, J.C.; Marín, M.C. Regulatory feedback loop between TP73 and TRIM32. Cell Death Dis. 2013, 4, e704.
- Peschiaroli, A.; Scialpi, F.; Bernassola, F.; Pagano, M.; Melino, G. The F-box protein FBXO45 promotes the proteasome-dependent degradation of p73. Oncogene 2009, 28, 3157–3166.
- Malatesta, M.; Peschiaroli, A.; Memmi, E.M.; Zhang, J.; Antonov, A.V.; Green, D.R.; Barlev, N.A.; Garabadgiu, A.V.; Zhou, P.; Melino, G.; et al. The Cul4A–DDB1 E3 ubiquitin ligase complex represses p73 transcriptional activity. Oncogene 2012, 32, 4721–4726.
- Chaudhary, N.; Maddika, S. WWP2-WWP1 ubiquitin ligase complex coordinated by PPM1G maintains the balance between cellular p73 and ΔNp73 levels. Mol. Cell. Biol. 2014, 34, 3754–3764.
- Sayan, B.S.; Yang, A.L.; Conforti, F.; Tucci, P.; Piro, M.C.; Browne, G.J.; Agostini, M.; Bernardini, S.; Knight, R.A.; Mak, T.W.; et al. Differential control of TAp73 and DeltaNp73 protein stability by the ring finger ubiquitin ligase PIR2. Proc. Natl. Acad. Sci. USA 2010, 107, 12877–12882.
- Balint, E.; Bates, S.; Vousden, K.H. Mdm2 binds p73 alpha without targeting degradation. Oncogene 1999, 18, 3923–3929.
- Zeng, X.; Chen, L.; Jost, C.A.; Maya, R.; Keller, D.; Wang, X.; Kaelin, W.G.; Oren, M.; Chen, J.; Lu, H. MDM2 Suppresses p73 Function without Promoting p73 Degradation. Mol. Cell. Biol. 1999, 19, 3257–3266.
- Dobbelstein, M.; Wienzek, S.; König, C.; Roth, J. Inactivation of the p53-homologue p73 by the mdm2-oncoprotein. Oncogene 1999, 18, 2101–2106.
- Neira, J.L.; Díaz-García, C.; Prieto, M.; Coutinho, A. The C-terminal SAM domain of p73 binds to the N terminus of MDM2. Biochim. Biophys. Acta (BBA) Gen. Subj. 2019, 1863, 760–770.
- Wang, X.; Arooz, T.; Siu, W.Y.; Chiu, C.H.; Lau, A.; Yamashita, K.; Poon, R.Y. MDM2 and MDMX can interact differently with ARF and members of the p53 family. FEBS Lett. 2001, 490, 202–208.
- Hosoda, M.; Ozaki, T.; Miyazaki, K.; Hayashi, S.; Furuya, K.; Watanabe, K.-I.; Nakagawa, T.; Hanamoto, T.; Todo, S.; Nakagawara, A. UFD2a mediates the proteasomal turnover of p73 without promoting p73 ubiquitination. Oncogene 2005, 24, 7156–7169.
- Min, B.; Ryu, J.; Chi, S.-W.; Yi, G.-S. Ubiquitination-dependent degradation of p73 by the mitochondrial E3 ubiquitin ligase Hades. Biochem. Biophys. Res. Commun. 2015, 467, 316–321.
- Alghamdi, R.M.; Hassan, M.A.; Kaleem, M.; Kayali, A.; Halwani, M.A.; Zamzami, M.A.; Choudhry, H.; Alhosin, M. Targeting Itch/p73 pathway by thymoquinone as a novel therapeutic strategy for cancers with p53 mutation. Eur. J. Cell Sci. 2020, 2, 20–26.
- Luo, Z.-L.; Luo, H.-J.; Fang, C.; Cheng, L.; Huang, Z.; Dai, R.; Li, K.; Tian, F.-Z.; Wang, T.; Tang, L.-J. Negative correlation of ITCH E3 ubiquitin ligase and miRNA-106b dictates metastatic progression in pancreatic cancer. Oncotarget 2015, 7, 1477–1485.
- Bernassola, F.; Karin, M.; Ciechanover, A.; Melino, G. The HECT Family of E3 Ubiquitin Ligases: Multiple Players in Cancer De-velopment. Cancer Cell 2008, 14, 10–21.
- Oberst, A.; Malatesta, M.; Aqeilan, R.I.; Rossi, M.; Salomoni, P.; Murillas, R.; Sharma, P.; Kuehn, M.R.; Oren, M.; Croce, C.M.; et al. The Nedd4-binding partner 1 (N4BP1) protein is an inhibitor of the E3 ligase Itch. Proc. Natl. Acad. Sci. USA 2007, 104, 11280–11285.
- Hou, H.; Sun, D.; Zhang, X. The role of MDM2 amplification and overexpression in therapeutic resistance of malignant tumors. Cancer Cell Int. 2019, 19, 1–8.
- Sznarkowska, A.; Maleńczyk, K.; Kadziński, L.; Bielawski, K.P.; Banecki, B.; Zawacka-Pankau, J. Targeting of p53 and its homolog p73 by protoporphyrin IX. FEBS Lett. 2010, 585, 255–260.
- Duan, W.; Gao, L.; Wu, X.; Zhang, Y.; Otterson, G.A.; Villalona-Calero, M.A. Differential response between the p53 ubiquitin–protein ligases Pirh2 and MdM2 following DNA damage in human cancer cells. Exp. Cell Res. 2006, 312, 3370–3378.
- Sheng, Y.; Laister, R.C.; Lemak, A.; Wu, B.; Tai, E.; Duan, S.; Lukin, J.A.; Sunnerhagen, M.; Srisailam, S.; Karra, M.D.; et al. Molecular basis of Pirh2-mediated p53 ubiquitylation. Nat. Struct. Mol. Biol. 2008, 15, 1334–1342.
- Yang-Hartwich, Y.; Tedja, R.; Roberts, C.M.; Goodner-Bingham, J.; Cardenas, C.; Gurea, M.; Sumi, N.J.; Alvero, A.B.; Glackin, C.A.; Mor, G. p53-Pirh2 Complex Promotes Twist1 Degradation and Inhibits EMT. Mol. Cancer Res. 2019, 17, 153–164.
- Wu, H.; Leng, R.P. UBE4B, a ubiquitin chain assembly factor, is required for MDM2-mediated p53 polyubiquitination and deg-radation. Cell Cycle 2011, 10, 1912–1915.
- Wu, X.; Sha, J.; Liu, D.; Chen, Y.; Yang, G.; Zhang, J.; Bo, J.; Huang, Y.; Chen, Y. High expression of P53-induced Ring-h2 protein is associated with poor prognosis in clear cell renal cell carcinoma. Eur. J. Surg. Oncol. (EJSO) 2013, 39, 100–106.
- Jung, Y.S.; Qian, Y.; Chen, X. The p73 tumor suppressor is targeted by Pirh2 RING finger E3 ubiquitin ligase for the pro-teasome-dependent degradation. J. Biol. Chem. 2011, 286, 35388–35395.
- Horn, E.J.; Albor, A.; Liu, Y.; El-Hizawi, S.; VanderBeek, G.E.; Babcock, M.; Bowden, G.T.; Hennings, H.; Lozano, G.; Weinberg, W.C.; et al. RING protein Trim32 associated with skin carcinogenesis has anti-apoptotic and E3-ubiquitin ligase properties. Carcinogenesis 2003, 25, 157–167.
- Kano, S.; Miyajima, N.; Fukuda, S.; Hatakeyama, S. Tripartite Motif Protein 32 Facilitates Cell Growth and Migration via Degradation of Abl-Interactor 2. Cancer Res. 2008, 68, 5572–5580.
- Boominathan, L. The guardians of the genome (p53, TA-p73, and TA-p63) are regulators of tumor suppressor miRNAs network. Cancer Metastasis Rev. 2010, 29, 613–639.
- Lin, M.; Wang, Z.-W.; Zhu, X. FBXO45 is a potential therapeutic target for cancer therapy. Cell Death Discov. 2020, 6, 55.
- Wang, K.; Qu, X.; Liu, S.; Yang, X.; Bie, F.; Wang, Y.; Huang, C.; Du, J. Identification of aberrantly expressed F-box proteins in squamous-cell lung carcinoma. J. Cancer Res. Clin. Oncol. 2018, 144, 1509–1521.
- Kogure, N.; Yokobori, T.; Ogata, K.; Altan, B.; Mochiki, E.; Ohno, T.; Toyomasu, Y.; Yanai, M.; Kimura, A.; Yanoma, T.; et al. Low Expression of FBXO45 Is Associated with Gastric Cancer Progression and Poor Prognosis. Anticancer Res. 2017, 37, 191–196.
- Chen, X.; Sahasrabuddhe, A.A.; Szankasi, P.; Chung, F.; Basrur, V.; Rangnekar, V.M.; Pagano, M.; Lim, M.S.; Elenitoba-Johnson, K.S.J. Fbxo45-mediated degradation of the tumor-suppressor Par-4 regulates cancer cell survival. Cell Death Differ. 2014, 21, 1535–1545.
- Richter, K.T.; Kschonsak, Y.T.; Vodicska, B.; Hoffmann, I. FBXO45-MYCBP2 regulates mitotic cell fate by targeting FBXW7 for degradation. Cell Death Differ. 2019, 27, 758–772.
- Mahoney, J.A.; Odin, J.A.; White, S.M.; Shaffer, D.; Koff, A.; Casciola-Rosen, L.; Rosen, A. The human homologue of the yeast polyubiquitination factor Ufd2p is cleaved by caspase 6 and granzyme B during apoptosis. Biochem. J. 2002, 361, 587–595.
- Wu, H.; Pomeroy, S.L.; Ferreira, M.; Teider, N.; Mariani, J.; Nakayama, K.I.; Hatakeyama, S.; Tron, V.A.; Saltibus, L.F.; Spyracopoulos, L.; et al. UBE4B promotes Hdm2-mediated degradation of the tumor suppressor p53. Nat. Med. 2011, 17, 347–355.
- Ma, Y.; Yu, W.-D.; Hershberger, P.A.; Flynn, G.; Kong, R.-X.; Trump, N.L.; Johnson, C.S. 1α,25-Dihydroxyvitamin D3 potentiates cisplatin antitumor activity by p73 induction in a squamous cell carcinoma model. Mol. Cancer Ther. 2008, 7, 3047–3055.
- Chatterjee, A.; Upadhyay, S.; Chang, X.; Nagpal, J.K.; Trink, B.; Sidransky, D. U-box-type ubiquitin E4 ligase, UFD2a attenuates cisplatin mediated degradation of DeltaNp63alpha. Cell Cycle 2008, 7, 1231–1237.
- Jung, J.H.; Bae, S.; Lee, J.Y.; Woo, S.R.; Cha, H.J.; Yoon, Y.; Suh, K.S.; Lee, S.J.; Park, I.C.; Jin, Y.W.; et al. E3 ubiquitin ligase Hades negatively regulates the exonuclear function of p53. Cell Death Differ. 2011, 18, 1865–1875.
- Angers, S.; Li, T.; Yi, X.; MacCoss, M.J.; Moon, R.T.; Zheng, N. Molecular architecture and assembly of the DDB1–CUL4A ubiquitin ligase machinery. Nat. Cell Biol. 2006, 443, 590–593.
- Higa, L.A.; Wu, M.; Ye, T.; Kobayashi, R.; Sun, H.; Zhang, H. CUL4–DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat. Cell Biol. 2006, 8, 1277–1283.
- Hu, J.; McCall, C.M.; Ohta, T.; Xiong, Y. Targeted ubiquitination of CDT1 by the DDB1–CUL4A–ROC1 ligase in response to DNA damage. Nat. Cell Biol. 2004, 6, 1003–1009.
- Nag, A.; Bagchi, S.; Raychaudhuri, P. Cul4A Physically Associates with MDM2 and Participates in the Proteolysis of p53. Cancer Res. 2004, 64, 8152–8155.
- Maddika, S.; Kavela, S.; Rani, N.; Palicharla, V.R.; Pokorny, J.L.; Sarkaria, J.N.; Chen, J. WWP2 is an E3 ubiquitin ligase for PTEN. Nat. Cell Biol. 2011, 13, 728–733.

