1000/1000
Hot
Most Recent
 +1 point
+1 point
Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant genetic disorder that presents with telangiectases in skin and mucosae, and arteriovenous malformations (AVMs) in internal organs such as lungs, liver, and brain. Mutations in ENG (endoglin), ACVRL1 (ALK1), and MADH4 (Smad4) genes account for over 95% of HHT. Localized telangiectases and AVMs are present in different organs, with frequencies which differ among affected individuals. By itself, HHT gene heterozygosity does not account for the focal nature and varying presentation of the vascular lesions leading to the hypothesis of a “second-hit” that triggers the lesions. Accumulating research has identified a variety of triggers that may synergize with HHT gene heterozygosity to generate the vascular lesions. Among the postulated second-hits are: mechanical trauma, light, inflammation, vascular injury, angiogenic stimuli, shear stress, modifier genes, and somatic mutations in the wildtype HHT gene allele.
Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant vascular disorder that exhibits age-related penetrance and extensive clinical variability, including intra-familial variability [1]. The characteristic vascular lesions range from 1 to 2 mm punctate mucocutaneous telangiectases to arteriovenous malformations (AVMs) several centimeters in diameter within visceral organs, particularly the lungs, liver, and brain. Telangiectases close to the surface of the skin and mucous membranes are fragile and frequently rupture and bleed upon slight trauma. Spontaneous and recurrent nose-bleeding (epistaxis) typically begins in mid-childhood and is the most common clinical manifestation; although occurring in over 90% of patients, the severity varies from an infrequent few drops to brisk bleeds multiple times daily. Gastrointestinal (GI) bleeding due to mucosal telangiectases affects approximately 25% of patients, almost always presenting after the age of 50. Many HHT patients have iron-deficiency anemia secondary to chronic bleeding of telangiectases, more often from nasal than GI lesions [2][3].
Solid organ AVMs are direct connections between artery and vein which bypass capillary beds and result in life-threatening complications more often related to the shunting of blood per se through these low resistance pathways, than to hemorrhage. For example, pulmonary AVMs (PAVMs), which occur in about 50% of HHT patients overall, result in high-flow continuous intrapulmonary right-to-left shunts with significant related risk for stroke or brain abscess [4][5]. The majority of PAVMs (70% or more) occur in HHT patients, but approximately 20% are acquired and associated with trauma, cardiothoracic surgery, hepatic cirrhosis, metastatic cancer, mitral stenosis, infection, amyloidosis, and chronic thromboembolic disease [6]. The frequency of hepatic vascular malformations was approximately 75% in two studies that systematically imaged the liver of affected individuals using computed tomography (CT) [7][8] and 41% in another study using ultrasound examination [9], although only a minority (8% in the CT study) were symptomatic. When symptomatic, hepatic vascular malformations associated with HHT typically present in later adulthood as high output heart failure, due to significantly increased blood flow through shunting pathways in the liver [10]. Intracranial hemorrhage is the risk posed by brain AVMs, which are typically congenital and occur in about 10% of those with HHT [11][12].
The clinical diagnosis of HHT is based on the Curaçao Criteria: (i) recurrent and spontaneous nosebleeds (epistaxis), (ii) cutaneous or mucosal telangiectases on the skin of the hands, lips, or face, or inside of the nose or oral cavity, (iii) visceral AVMs or telangiectases in one or more of the internal organs, including lungs, brain, liver, gastrointestinal tract, and spinal cord, and (iv) family history of HHT (i.e., first-degree relative with a definite HHT clinical or genetic diagnosis). The HHT diagnosis is considered definite if three criteria are present, possible or suspected with two criteria, and unlikely if only one is present [3][13]. It is of note that the disease experts who initially created and twenty years later confirmed these consensus clinical criteria for HHT, believe that the locations of telangiectases and AVMs are specific in HHT. In fact, multiple telangiectases or an AVM in a location other than those considered characteristic actually argue against the diagnosis of HHT.
Overall, the phenotype and age of onset of manifestations is highly variable in HHT and this variability seems to depend on HHT subtype, as well as genetic background and/or environmental triggers (second-hits) to which each individual is exposed.
Heterozygous mutations in several genes are known to cause HHT. Endoglin (ENG) mutations cause what is referred to as HHT1 (OMIM #187300) [14], activin receptor-like kinase 1 (ACVRL1) mutations cause HHT2 (OMIM #600376) [15], while mothers against decapentaplegic homolog 4 (MADH4 or SMAD4) mutations cause a syndrome which combines familial juvenile polyposis and HHT (JP/HT; OMIM #175050) [16]. Also, mutations in the GDF2 gene, encoding bone morphogenetic protein 9 (BMP9), were described as the cause of an HHT-like syndrome [17], also named as HHT5 (OMIM #615506). Two further loci have been found by linkage analyses on chromosomes 5 (HHT3) and 7 (HHT4), but their corresponding coding genes have not been identified [18][19].
ENG and ACVRL1 are the predominant genes mutated in HHT, each responsible for almost half of cases. A mutation in one of these two genes is detected in over 95% of individuals who meet Curaçao diagnostic criteria, and a mutation in SMAD4 is detected in an additional 1–2% [20][21]. Although the phenotypes generated by mutations in ENG or ACVRL1 are similar enough that they cannot be reliably distinguished in the clinical setting, pulmonary and cerebral AVMs are more frequent in HHT1 patients while GI bleeding and liver AVMs are more common in HHT2 [22][23]. Studies suggest that solid organ AVMs in JP/HHT are at least as common as in HHT1 and HHT2, and that pulmonary AVMs may be more frequent [16][21][24][25]. Overall, Curaçao diagnostic criteria for HHT are highly predictive of a pathogenic variant in ENG (HHT1) or ACVRL1 (HHT2) but cannot distinguish between these two genotypes [21]. Furthermore, the genetic heterogeneity does not explain the striking variable expression observed within families.
There are no common disease-causing mutations or mutation hot spots in any of the HHT genes. Both ENG and ACVRL1 mutations that cause HHT are dispersed almost equally throughout the genes except in a couple of exons. Mutations of all types have been reported [20][26]. The frequency of single or several exon deletions/duplications is up to 10%. Mutations causing sequence changes are slightly more common in the ENG than ACVRL1. Missense mutations account for more than half of mutations detected, however, nonsense, deletions, insertions, and splice site mutations have also been reported. Mutant ENG and ACVRL1 proteins, including the products of multiple missense mutations, were shown to be expressed at the 50% level, in accordance to the haploinsufficiency model [20].
All the genes mutated in HHT encode proteins involved in the signaling pathway of the transforming growth factor beta (TGF-β) superfamily, including bone morphogenetic proteins (BMPs) (Figure 1). The most likely affected pathway in HHT involves the auxiliary receptor endoglin associated with the signaling serine/threonine kinase receptor ALK1. Both proteins are able to bind the ligands BMP9 and BMP10 [27][28][29], which form a heterodimeric complex that provides most of their BMP biological activity in plasma [30][31]. Upon ligand binding, ALK1 phosphorylates Smad1/5/8 followed by their nuclear translocation in complex with Smad4 [32][33][34][35]. Because endoglin and ALK1 are predominantly expressed in endothelial cells, which respond to circulating BMP9, they are widely accepted as the target cells most affected in HHT.
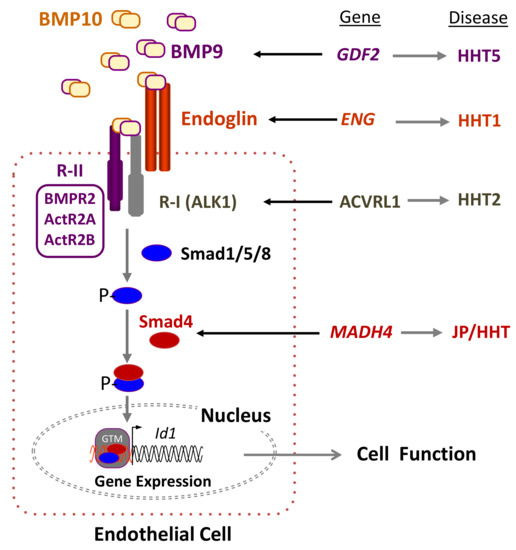
Figure 1. Hereditary hemorrhagic telangiectasia (HHT) and the transforming growth factor beta (TGF-β) signaling pathway in endothelial cells. Heterodimers of bone morphogenetic protein 9 (BMP9) and BMP10, among other members of the TGF-β family, bind to an endothelial cell surface receptor complex composed by the type I (R-I) receptor named ALK1 and the type II (R-II; BMPR2, ActR2A, ActR2B) receptor, both serine/threonine kinases, as well as the auxiliary receptor endoglin. The heterodimeric association between different R-I and R-II determines the specificity of the ligand signaling. Upon ligand binding, the R-II transphosphorylates ALK1, which subsequently propagates the signal by phosphorylating the receptor-regulated Smad (R-Smad) family of proteins, Smad1/5/8. Once phosphorylated (P-), R-Smads form heteromeric complexes with a cooperating homologue named Smad4 and translocate into the nucleus, where they regulate the transcriptional activity of different target genes, in turn modulating endothelial cell function. The involvement of other components of the TGF-β pathway has been omitted for simplification [32]. BMP9, Endoglin, ALK1, and Smad4 proteins are encoded by GDF2, ENG, ACVRL1, and MADH4 genes, whose pathogenic mutations give rise to HHT5, HHT1, HHT2, and JPHT, respectively. BMP, bone morphogenetic protein; GTM, general transcription machinery. Adapted from Ruiz-Llorente et al. [34].
It is worth noting that within the TGF-β system, endoglin is known to participate in several receptor complexes, not necessarily including ALK1, and is capable of binding different ligands [36][37][38][39]. In addition, endoglin has been reported to be involved in several pathways relevant to vascular homeostasis. Endoglin was shown to regulate the organization of the actin cytoskeleton in endothelial cells via the interaction of its cytoplasmic domain with zyxin and zyxin-related protein 1 (ZRP1) [40][41]. Upon interacting with these members of the zyxin family, endoglin coordinates matrix-dependent cues with actin dynamics like stress fibers and focal adhesions. In primary cultures of endothelial cells from HHT1 patients, endoglin deficiency appears to lead to a disorganized F-actin cytoskeleton and abnormal tube formation [42]. This is in agreement with the fragile mucocutaneous telangiectases that easily break, leading to the frequent nose and gastrointestinal bleedings present in HHT patients. An active search for novel endoglin-specific interactors has allowed the identification of multiple proteins, suggesting the involvement of endoglin in ALK1-independent pathways; nonetheless, the functional characterization and relevance of the novel interactors to the HHT1 field remains to be established [39][43]. Overall, it can be postulated that the phenotypic differences between HHT1 and HHT2 could arise, at least in part, from a subthreshold endoglin participation in these ALK1-independent signaling pathways.
A deficient expression of the HHT genes underlies the molecular basis of disease pathogenesis. Mono-allelic loss of expression leading to haploinsufficiency of the respective HHT1 and HHT2 proteins has been shown to dysregulate TGF-β/BMP signaling in endothelial cells negatively impacting cell proliferation, migration, and recruitment during vascular remodeling and angiogenesis [20][34][44][45].
Different Eng or Acvrl1 genetic mouse models of HHT have been described by several groups during the last three decades [45]. Mice lacking functional endoglin [46][47][48] or ALK1 [49][50] were generated by germline gene-targeting. In all cases, global embryonic loss of endoglin or ALK1 expression leads to cardiovascular defects, enlarged fragile vessels, and embryonic lethality by mid-gestation. These early models suggested that both endoglin and ALK1 were essential for cardiovascular development and homeostasis and that a further decrease in the level of these essential proteins could lead to vascular abnormalities.
The second-hit hypothesis, also known as Knudson hypothesis, was first described to explain the progression of cancer [51]. It was suggested that the first event of tumorigenesis in familiar cancers would be due to germline inactivation of one allele, followed by somatic inactivation of the second allele. Recently, using next-generation sequencing, Snellings et al. [52] have been able to demonstrate the presence of low-frequency somatic mutations in telangiectases of HHT1 and HHT2 patients, suggesting that the bi-allelic loss of ENG or ACVRL1 was required for the development of vascular lesions. Overall, haploinsufficiency is widely accepted as the underlying cause of HHT1 and HHT2 pathogenicity [20][34][53], although a dominant negative effect has been described in a few ENG and ACVRL1 pathogenic mutations [54][55][56][57]. Nonetheless, neither haploinsufficiency nor a dominant negative effect by itself can account for the localized generation of vascular lesions in HHT patients. It is intriguing that the vascular HHT lesions appear only at distinct sites within certain organs, rather than being present throughout the body and in all organs/tissues. This paradox has been explained, as in many other genetic diseases, by postulating the need for a second-hit.
The second-hit hypothesis has evolved over the years in order to explain how multiple factors, either environmental or genetic (modifier genes or somatic mutations), can contribute to complex diseases with phenotypic heterogeneity. In the case of HHT, external or physiological triggers such as vascular injury, inflammation, and angiogenic stimuli could account for the generation of AVMs. We will first describe the environmental factors proposed for HHT and their effects on endothelial cell functions. We will then describe how modifier genes or a somatic mutation in the normal HHT allele may synergize with a background of deficient endoglin or ALK1 expression/activity to generate vascular lesions [58] (Figure 2).
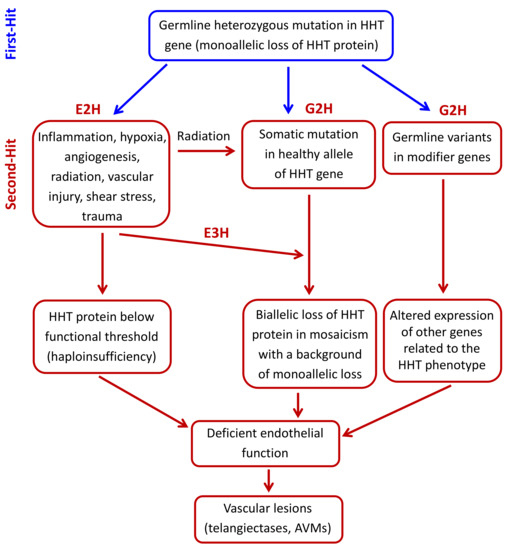
Figure 2. Hypothetical second-hit model in hereditary hemorrhagic telangiectasia (HHT). The germline heterozygous mutation in the HHT gene leads to a monoallelic loss of the encoded HHT protein in endothelial cells (First-hit). A subsequent environmental stimulus like inflammation, hypoxia, neoangiogenesis, vascular injury, radiation, shear stress, or trauma (environmental second-hit; E2H), can induce the expression/activation of mediators, which generate a microenvironment where HHT protein levels are below the needed functional threshold. This drop in the HHT functional protein can also be generated by a somatic mutation in the normal allele (genetic second-hit; G2H), leading to a focal protein loss in lesions. One possible cause of somatic mutation is sunlight radiation, especially in skin telangiectases. A somatic mutation could also synergize with an environmental third-hit (E3H). Modifier genes (G2H) could also contribute to focal vascular lesions by affecting HHT protein level and activity. In all cases, the result is an impaired endothelial cell function, leading to the generation of telangiectases or arteriovenous malformations (AVMs).
The two-hit hypothesis has been widely accepted for many years as a plausible explanation for the phenotypic variability found in HHT. Here, we have reviewed some of the reported environmental and genetic second-hits (Figure 2). Thus, germline heterozygous mutations in HHT genes (first-hit) result in a monoallelic protein loss in endothelial cells. A subsequent environmental stimulus (second-hit) like inflammation, hypoxia, neoangiogenesis, vascular injury, shear stress, radiation, or trauma, can induce the expression/activation of mediators, which generate a microenvironment where HHT protein levels are below the needed functional threshold. Furthermore, the existence of a genetic second-hit (somatic mutation in the normal HHT allele), combined with an environmental trigger (tertiary-hit), and/or the presence of modifier variants, can set a much lower threshold for disease development. In all cases, the consequence is an impaired endothelial cell function that leads to the generation of telangiectases and AVMs. Overall, the current clinical data, as well as in vivo and in vitro experimental results, support the second-hit hypothesis to explain why certain individuals with HHT genotypes develop earlier and/or more severe clinical phenotypes than other family members. Also, the organ-specific location of telangiectases and AVMs in HHT, and the differing age of presentation for lesions at each site, suggest that the required second-hits may be tissue-specific. However, major gaps in our knowledge remain, particularly in delineating the exact role of the different second-hits that lead to the development of clinically relevant symptoms in HHT, and how this information can be applied in the clinical practice. Further studies to better understand the pathological mechanisms of HHT, including identification of novel potential second-hits, are needed, as well as the translation of this knowledge into preventive clinical measures and treatments.




