1000/1000
Hot
Most Recent
 +1 point
+1 point
DNA double-strand breaks (DSBs) have been recognized as the most serious lesions in irradiated cells. While several biochemical pathways capable of repairing these lesions have been identified, the mechanisms by which cells select a specific pathway for activation at a given DSB site remain poorly understood. The impact of chromatin and repair foci architecture on these mechanisms can be elucidated by super-resolution microscopy in combination with mathematical approaches of topology. These aspects are discussed in relation to state of the art knowledge of ionizing radiation induced damaging of cell nuclei and DNA repair.
Double-strand breaks (DSBs) are the most deleterious type of DNA lesion and are induced in DNA by ionizing radiation, radiomimetic chemicals and cellular processes [1][2][3]. Theoretically, a single DSB may lead to cell death or initiate carcinogenesis if left unrepaired or repaired improperly [4]. After exposure to high doses of sparsely ionizing radiation or even low doses of densely ionizing radiation, there is a serious risk that numerous and possibly clustered DSBs will not be repaired in a timely manner, leading to separation of broken DNA ends, misrejoining of these ends, and formation of often lethal chromosomal aberrations. These events probably explain why fast repair mechanisms have evolved and are preferred by organisms with large genomes [5]. However, a fast rate of repair may be at the expense of repair accuracy, resulting in smaller mutations, some of which may be carcinogenic and thus no less dangerous than larger mutations. Hence, damaged cells have to solve a serious repair dilemma and maintain a careful balance between repair speed and fidelity.
In mammals, the two main repair pathways with these opposite repair strategies are the fast but error-prone nonhomologous end joining (NHEJ) and the much slower but usually precise homologous recombination (HR) [6]. Unsurprisingly, NHEJ and HR utilize, in principle, different repair mechanisms (Figure 1A) specialized to cope with different repair targets and scenarios. In addition, alternative repair pathways (hereafter and in the figures collectively referred to as alternative end joining; A-Ej) have been identified (Figure 1A), which extend or back up the conventional repair pathways in situations that remain incompletely understood [7][8][9][10][11][12]. These pathways combine aspects of both NHEJ and HR mechanisms to various degrees [13], as reflected in their problematic and still inconsistent categorization. Most often reported are alternative NHEJ (aNHEJ; also known as backup NHEJ, bNHEJ), single-strand annealing (SSA), and microhomology-mediated end joining (MMEJ), which differ in the requirement for some repair proteins, extent of DNA end resection, and length of homology needed for recombination [7][14]. NHEJ and HR always offer—because of their opposite advantages and disadvantages—only a compromise solution, indicating the requirement for precise regulation of mutual repair pathway competition and cooperation within the repair network.

Figure 1. Schematic representation of prominent pan-nuclear-acting (global) factors, global factors acting randomly at different sites, and site-specific (local) factors that participate in the selection of DSB repair pathways at individual DSB damage sites. (A) Left: definition of the nuclear competence of repair pathway-selecting factor types; the area of competence is indicated by the red frames. Right: DSB repair pathways plus their principles and mutual transitions depending on the cell cycle phase (G1 vs. S/G2 cells). (B) Examples of global factors (a–d) having a pancellular effect on DSB repair pathways and their selection. Repair pathways preferred or affected by each of these factors and the character of their influence are suggested. (C) The relationship between three interdependent factors related to irradiation that have a global mode of action but locally specific effects—radiation LET, irradiation conditions (dose, dose rate) and chromatin architecture (a–c)—is proposed, together with the potential outcomes of these factors on DSB repair pathway selection. (D) Diversity of radiation-induced DSB damage sites in terms of (a) the characteristics of broken DNA ends, the architecture and function of damaged chromatin (b), and the epigenetic code. The influence of these local factors on DSB repair pathways is indicated. For interactions between factors B, C and D and their joint effect on the activation of particular DSB repair pathways.
Microscopic research of DSB repair is further complicated by the fact that some proteins of which only a few molecules are needed, do not form extended, microscopically distinguishable ionizing radiation induced foci (IRIFs) and can thus be visualized only by superresolution visualization methods. Electron microscopy has yielded surprising results regarding the focal accumulation of repair proteins in euchromatin and heterochromatin. Lorat et al. [15][16], who analyzed the nuclear distribution of various repair proteins in cells irradiated with low-LET and high-LET radiation showed that at two time periods post irradiation (0.5 and 5 h), γH2AX, MDC1, and 53BP1 can be detected only in heterochromatin domains positive for H3K9me3, while the Ku70/80 heterodimer can be detected in both euchromatin and heterochromatin. This observation strongly suggests the involvement of the micro- and/or nanoarchitecture of chromatin and, subsequently, IRIFs in the selection and/or propagation of a particular repair pathway. However, the absence of the indicated protein foci in euchromatin has not been confirmed by any other technique and contradicts the results of confocal microscopy.
Methodologically, this contradiction may be explained by a gap in resolution and thus a gap in knowledge in the 50 nm to 200 nm scale range. Whereas the strength of electron microscopy lies in the low-10-nm scale range, optical confocal microscopy covers resolutions above 200 nm. Thus, for decades during the second half of the 20th century, the scale range between electron and confocal microscopy, although highly relevant for biomolecular dynamics, seemed to be obscured for gaining scientific insights. Therefore, great hopes are currently placed in emerging studies using superresolution light microscopic techniques [17][18], which cover this critical gap of visualization and approach a resolution of 10 nm while preserving the advantages of optical microscopy.
We recently introduced single-molecule localization microscopy (SMLM) [19][20] to simultaneously analyze the architecture of damaged chromatin domains and IRIFs at the nanoscale (see, for example, Figure 1; compare the widefield image, panel A, with SMLM images, panels B–D) [21][22][23]. SMLM is one of the superresolution (nanoscopy) techniques established in recent decades [24]. In addition to having an improved resolution of approximately 10 nm, SMLM is renowned for providing quantitative data on 2D/3D localization (coordinates) and other signal parameters of individual molecules of interest without a need for complicated image analysis. Several SMLM and other nanoscale studies have shown that IRIFs have an internal nanoarchitecture, with nanoclusters of γH2AX and individual proteins occupying nonoverlapping space [25][26].
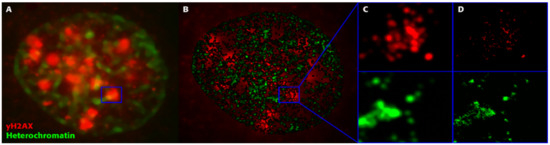
Figure 1. Superresolution imaging of a breast cancer (SkBr3) cell during DSB repair after exposure to 1 Gy X-rays. From such images, characteristic molecular arrangements during repair are elucidated. (A) Overview image acquired by widefield microscopy. The blue square (2 µm × 2 µm) encloses a typical γH2AX focus. (B) Superresolution SMLM image of heterochromatin (green) and γH2AX foci (red), with the γH2AX overview image in the background indicating the reduced z-slice depth in the SMLM image reconstructed from the label point coordinates. (C) Magnification of the marked region (2 µm × 2 µm) in the SMLM image with Gaussian blur but without the background image; the two color channels are separated in the upper and lower images. (D) The same image as (C) but with maximum precision of label points (each point corresponds to a single fluorescent molecule of the indicated antibody). Maximum precision means the highest image resolution that can be obtained from the SMLM data set. (Note: In all images, the blue squares enclose an area of 2 µm × 2 µm and can be used as scale bars.).
With the SMLM data matrix of molecule coordinates, Ripley’s metrics for pairwise distance frequency histograms [23][27] can be applied to evaluate structures, molecular clusters, or spatial distributions of label points and their dynamic rearrangements during repair (Figure 2; compare panels A and B for 500 mGy and 4 Gy exposure to X rays) [21][25][28]. Using these approaches of structure elucidation from distance frequency patterns, together with newly developed mathematical topological tools based on persistent homology [29], we showed (for SkBr3 cells exposed to 1 Gy X-rays) that the topological similarity and, thus, the nanoarchitecture of γH2AX clusters depends on the distance of the clusters from heterochromatin (Figure 1) [21]. High topological similarities were also found for 53BP1 clusters in repair foci along high-LET 15N particle tracks in neonatal human dermal fibroblasts (NHDF) and the U87 glioblastoma cell line [30]. More generally, this finding means that the architecture of γH2AX and 53BP1 clusters is not random and depends on the chromatin environment at DSB sites, consistent with the results of high-resolution ChIP-seq mapping of γH2AX spreading from multiple DSBs induced at annotated positions in human DIvA cells [31]. This finding shows that phosphorylation follows a highly stereotyped pattern governed by the original (predamage) chromatin architecture. Provided that the chromatin architecture dictates γH2AX spreading, it is reasonable to suppose that the architecture of nascent γH2AX foci subsequently affects downstream repair events. Such events could be the binding and organization of repair proteins (such as MDC1 and 53BP1) to IRIFs, the insertion of epigenetic marks (e.g., ubiquitin) into IRIFs, and, in turn, the determination of the architecture of maturating or already dissolving IRIFs [32].
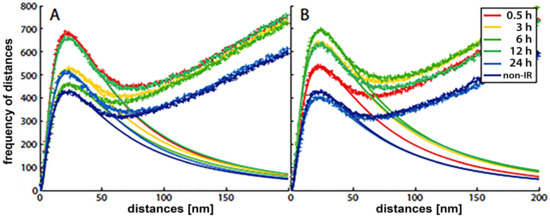
Figure 2. Frequency histograms of pairwise distances of H3K9me3 heterochromatin label points in breast cancer cell (SkBr3) nuclei at different times post irradiation with two doses of X-rays: (A) 500 mGy, (B) 4 Gy. The distributions of the crosses represent the experimentally measured results. The smooth curves, which follow a logarithmic Gaussian distribution, are fitted curves of the peaks below 100 nm, indicating cluster formation in heterochromatin. According to Ripley’s interpretation, the linearly increasing experimental curves describe a random behavior of molecule positions, i.e., the dense clusters are embedded in an environment of randomly, less densely arranged H3K9me3 marks. Non-IR: the nonirradiated control.
Indeed, the roles of numerous proteins in DSB repair dramatically depend on the specific conditions. The 53BP1 protein generally inhibits resection and promotes NHEJ [33]. However, at some DSB substrates, it shows the opposite effects. For instance, it stimulates resection and switches NHEJ to MMEJ [8][11][34]. In addition, 53BP1 enhances repair fidelity independent of the repair pathway [35]. Hence, 53BP1 and some other proteins, such as BRCA1, probably establish structural platforms that support the recruitment and assembly of the repair machinery in specific ways, dictated by integrated information from multiple global and local factors (reviewed in [36]). Strikingly, 53BP1 and RIF1 were only recently discovered to form an autonomous functional module that stabilizes three-dimensional chromatin topology at sites of DNA breakage [35].
Our SMLM analysis also revealed that the nanoarchitecture of γH2AX foci in heterochromatin shows a higher mutual similarity than γH2AX foci in euchromatin. These greater differences between IRIFs in euchromatin probably reflect the variability in the expression intensity across euchromatin loci, in contrast to the rather uniformly silenced heterochromatin. On the other hand, heterochromatin experiences especially extensive architectural reorganization associated with repair initiation and progression. Thus, γH2AX foci in euchromatin may still reflect the variable original architectures of differently expressed genomic domains, but the architecture of γH2AX foci in heterochromatin has already adopted the features of remodeling. Our results thus suggest that remodeling processes at different sites in heterochromatin broadly follow the same principles, indicating that the same repair mechanism is active across these sites. This situation is in contrast to the variable repair of DSBs in structurally and functionally heterogeneous euchromatin.
In addition, using SMLM, we showed that the formation kinetics and architecture of 53BP1 foci differ for normal (nontransformed) and tumor cells, represented in the study by human dermal fibroblasts and highly radioresistant U87 glioblastoma cells, respectively [30]. The data currently being processed seem to suggest that γH2AX, RAD51, MRE11 and potentially other repair proteins also form IRIFs with cell type-specific kinetics and architecture. These differences might contribute to differences between the cells in repair pathway utilization and capacity.
Other breakthrough studies supporting the idea that IRIF nanoarchitecture reflects the repair mechanism or even significantly contributes to the repair pathway choice were published by Reindl et al. [37][38]. By using STimulated Emission Depletion (STED) microscopy [39], another well-established superresolution fluorescence microscopy technique, the authors showed that two nanoscale subzones exist within HR- but not NHEJ-associated IRIFs. Specifically, the resection zone and the zone of surrounding modified chromatin were recognized in [37][38] and in our preliminary unpublished analyses. Furthermore, IRIFs formed by different repair proteins that have specific functions in the NHEJ and HR pathways, such as 53BP1, BRCA1 and RAD51, were clearly shown to have different architectures ([37][38] and our unpublished results). Finally, mutual reorganization of 53BP1, BRCA1 and RAD51 proteins in the frame of IRIFs correlated with switching between NHEJ and HR [37][38]. Hence, the HR, NHEJ, and perhaps A-Ej pathways seem to form IRIFs with characteristic architectures.
Several other studies on IRIF nanoarchitecture have recently been published but cannot be discussed here due to space limitations. However, these studies focus on IRIF ultrastructure rather than the relationship of this ultrastructure to the selection of repair pathways [40]. Future experiments on synchronized cells, cells with altered/manipulated DSB repair pathways, or cells exposed to high-LET ions, as discussed below, are expected to provide more accurate insights into the relationship between the repair mechanisms and nanoarchitecture of particular chromatin domain types and IRIFs.





