1000/1000
Hot
Most Recent
 +1 point
+1 point
A sharp increase in the permeability of the mitochondrial inner membrane known as mitochondrial permeability transition (or mPT) occurs in mitochondria under the conditions of Ca2+ and ROS stress. Permeability transition can proceed through several mechanisms. The most common mechanism of mPT is based on the opening of a cyclosporine A (CSA)-sensitive protein channel in the inner membrane. In addition to the CSA-sensitive pathway, mPT can occur through the transient opening of lipid pores, emerging in the process of formation of palmitate/Ca2+ complexes. This pathway is independent of CSA and likely plays a protective role against Ca2+ and ROS toxicity. The review considers molecular mechanisms of formation and regulation of the palmitate/Ca2+-induced pores, which we designate as PA-mPT to distinguish it from the classical CSA-sensitive mPT.
Ca2+ is an ion involved in the regulation of many processes within living organisms and cells, including muscle contraction, neurotransmission, regulation of energy metabolism, and many others. The key regulatory role of Ca2+ is linked to its ability to be selectively redistributed across biological membranes via tightly regulated selective channels, exchangers, and pumps in response to specific stimuli. In addition to its regulatory role in physiological processes, Ca2+ plays a critical role as a mediator of stress-induced cell death. In many pathologies, disorders of ion homeostasis lead to a rise of the Ca2+ concentration in the cytoplasm and mitochondria, which eventually triggers a sharp increase in the permeability of mitochondrial membranes and cell death.
Under stress conditions, the Ca2+-induced increase in the permeability of the inner mitochondrial membrane is caused by the opening of a large protein channel, the so called mitochondrial permeability transition pore (or mPTP). Many laboratories worldwide extensively study mPTP (see [1][2][3] for recent reviews). Traditionally, mPTP is described in the literature as a large pore which is located in the inner mitochondrial membrane and allows passage of molecules up to 1.5 kDa in size. The key inducers of mPTP are Ca2+ and ROS, but it can also be affected and modulated by many other agents. A specific feature of mPTP is its sensitivity to cyclosporine A (CSA). In most cases, mPTP opening causes depolarization of mitochondria and cell death. On the other hand, mPTP has also been suggested to play an important physiological role—in cases when it is activated in a specific mode: either low-conductance or transient (flickering) [4][5]. The molecular architecture of mPTP is not completely understood, but it is generally agreed that mPTP has a protein nature and its opening can be mediated by ATP synthase and adenine nucleotide translocase [6][7][8][9]. The specific contribution of these proteins in the opening of mPTP is still debated though [10][11].
The studies on the involvement of fatty acids in the mitochondrial permeability transition began in the mid-90’s of the past century. Initially, we discovered that the lipid fraction of the ethanolic mitochondrial extract induced an ion channel activity in planar lipid bilayers (black lipid membranes, BLM) [12]. Further purification and analysis of the fraction revealed that it contained some kind of a “Ca2+ sensor”, a protein-free component with high affinity for Ca2+ (Kd in the sub-µM range). The affinity further increased at alkaline pH (pH = 8.0), and reconstitution of the component into BLM enabled the formation of non-selective ion channels upon the addition of Ca2+ [13][14]. We also found that the component was predominantly located in the mitochondrial contact sites, the membrane’s regions enriched with transport proteins [13][14].
Further examination of the purified hydrophobic Ca2+-binding component revealed that it was mostly composed of long-chain saturated fatty acids, predominantly palmitic and stearic. Later, we demonstrated that synthetic palmitic and stearic acids had roughly the same Ca2+ affinity as the hydrophobic mitochondrial extract. At the same time, other hydrophobic compounds available in native membranes (including unsaturated fatty acids, phospholipids, and cardiolipin) had Ca2+ affinity being 1–2 orders of magnitude lower as compared to the affinity of palmitic and stearic acids (Table 1). Thus, we concluded that the Ca2+-binding properties of the hydrophobic mitochondrial extract were determined by the presence of stearic and palmitic acids [13].
Table 1. Binding of various fatty acids and lipids 1.
| Lipids | Relative Ca2+ Binding |
|---|---|
|
Lauric acid (12:0) |
0.50 ± 0.03 |
|
Myristic acid (14:0) |
7.30 ± 0.25 |
|
Palmitic acid (16:0) |
83.00 ± 0.75 |
|
Stearic acid (18:0) |
100.00 |
|
Eicosanoic acid (20:0) |
73.00 ± 2.5 |
|
Docosanoic acid (22:0) |
44.00 ± 1.2 |
|
Lignoceric acid (24:0) |
15.00 ± 0.35 |
|
Palmitoleic acid (16:1) |
1.90 ± 0.08 |
|
Oleic acid |
5.70 ± 0.12 |
|
Linoleic acid (18:2) |
0.65 ± 0.04 |
|
Linoleinic acid (18:3) |
0.87 ± 0.05 |
|
Arachidonic acid (20:4) |
1.10 ± 0.05 |
|
1-Palmitoyl-lysophosphatidylcholine |
0.43 ± 0.02 |
|
1-Stearoyl-lysophosphatidylcholine |
0.47 ± 0.02 |
|
1-Lauroyl-lysophosphatidylcholine |
0.43 ± 0.01 |
|
Lysophosphatidylserine |
0.54 ± 0.03 |
|
1,2-Dipalmitoyl-sn-glycero-3-phosphatidylcholine |
0.40 ± 0.2 |
|
1,2-Dipalmitoyl-sn-glycero-3-phosphatidylethanolamine |
0.22 ± 0.01 |
|
1-Palmitoyl-sn-glycero-1-3-phosphatidylethanolamine |
0.76 ± 0.03 |
|
Palmitoil-CoA |
0.43 ± 0.02 |
|
Cardiolipin |
0.60 ± 0.03 |
|
L-α-phosphatidic acid |
19.50 ± 0.8 |
|
Cholesterol |
0.33 ± 0.01 |
|
Cerebrosides |
0.20 ± 0.01 |
|
Sphingomyelin |
0.30 ± 0.01 |
1 Five microliters of 2 mM solution of each lipid were applied to a Polyvinylidene difluoride (PVDF) membrane and binding of Ca2+ to the sample was estimated at pH 8.5 in the presence of 5 µM45 CaCl2. Ca2+ binding was measured in counts per minute. Stearic acid was taken as a reference to calculate the relative Ca2+ binding. Data are expressed in percentage and as the means ± SD of four experiments.
As shown in further experiments, the reconstitution of synthetic palmitic and stearic acids into BLM and the subsequent addition of Ca2+ induced a non-specific ionic conductance, which, in some cases, was accompanied by oscillations of ion currents (Figure 1a) [15]. A similar effect (Ca2+-induced permeabilization of membranes containing palmitic and stearic acids) was observed in the experiments with liposomes composed of phospholipids (Figure 1c) [15][16]. Palmitic and stearic acids were also found to cause a Ca2+-dependent permeability transition in the membranes of mitochondria (Figure 1b) isolated from various tissues (liver, heart, and brain) and in the membranes of erythrocytes (Figure 1d). It should be emphasized that, in case of mitochondrial membranes, the effect was CSA-independent, suggesting that the mechanism of membrane permeabilization was different from that of classical mPTP, which is known to be inhibited by CSA [17][18].
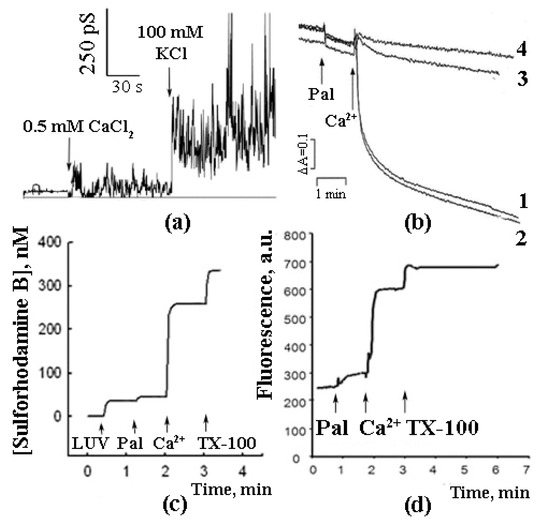
Figure 1. Palmitic acid can induce Ca2+-dependent permeabilization of artificial and biological membranes. (a) Planar lipid bilayer, PA 0.5%, membrane potential 100 mV. (b) Mitochondrial swelling: 1, 3, 4—no CSA, 2–1 µM CSA. Additions: (1, 2) 15 µM PA and 30 µM Ca2+, (3) 15 µM oleic acid (OA) and 30 µM Ca2+, (4) 15 µM brom-palmitic acid and 30 µM Ca2+. (c) Artificial liposcheme 50 µM PA and 1 mM Ca2+. (d) Plasmalemmal membranes of erythrocytes, release of the fluorescent probe sulforhodamine B from the erythrocytes induced by the addition of 50 µM PA and 1 mM Ca2+. Typical curves are shown (n = 6).
In the experiments with liposomes, membrane permeabilization was monitored by measuring the release of sulforhodamine B (SRB), a membrane-impermeable fluorescent dye. The amount of SRB released from liposomes was proportional to the amounts of palmitic acid (PA) and Ca2+ added [16]. The release of SRB occurred instantaneously and stopped abruptly (Figure 1b). The pH optimum of the effect was in the same range (pH = 8.0) as in the case of formation of PA/Ca2+ complexes [13]. Testing various fatty acids in these experiments revealed a pattern similar to that observed previously in the Ca2+ binding assays: (1) a bell-shaped dependence of the effect on the length of the hydrocarbon chain in the series of saturated fatty acids; (2) the maximal effect for palmitic and stearic acids, and (3) much lower efficiency of unsaturated fatty acids, such as linoleic acid (Figure 2).
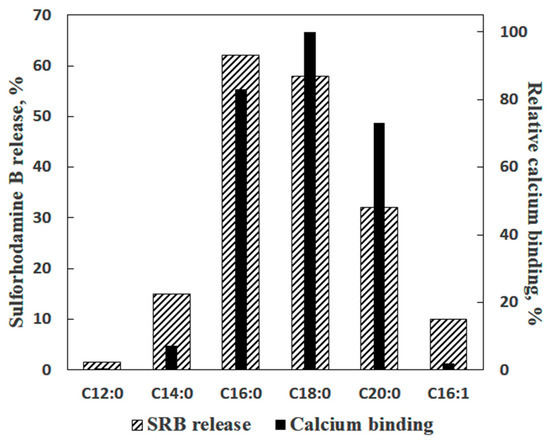
Figure 2. Ca2+-induced permeabilization of lecithin liposomes containing various fatty acids in comparison with the ability of fatty acids to bind Ca2+ ions. The data on the Ca2+-binding ability of fatty acids were taken from Table 1. Additions: 30 µM of a fatty acid and 1 mM Ca2+. The media contained 10 mM Tris-HCl (pH 8.5), 50 µM EGTA (ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid), and 40 µM KCl (n = 4).
The experiments with liposomes also showed that PA-containing vesicles could be permeabilized by other divalent cations (Ba2+, Mn2+, Ni2+, Co2+, and Sr2+, in the descending order). An “outlier” was Mg2+, whose effect was much lower than that of other divalent cations (twice as low as the effect of Sr2+ and more than thrice as low as the effect of Ca2+). Additionally, we confirmed, using fluorescent correlative spectroscopy, that SRB release was not caused by vesicle micellization and, therefore, should be attributed to the formation of pores in the liposomal membranes [16].
As can be seen in Figure 1, the combined effect of PA and Ca2+ (i.e., their ability to permeabilize the lipid bilayer) was demonstrated on both artificial and biological (mitochondrial and erythrocytic) membranes.
In case of intact mitochondria, the addition of PA alone did not produce any detectable effect, while the subsequent addition of Ca2+ induced a high-amplitude swelling of the organelles, indicating a transition in the permeability of the inner mitochondrial membrane to osmotic agents. This effect was not inhibited by CsA (Figure 1b) [13][17][19]. In the presence of PA and Ca2+, mitochondria became slightly depolarized but were able to maintain their function for quite a long time. We hypothesized that this phenomenological picture is characteristic of a flickering mode of permeability transition. The addition of EGTA or ruthenium red (i.e., the removal of Ca2+ or prevention of its uptake by mitochondria) resulted in the complete recovery of the membrane potential (Figure 3) [18], indicating that the flickering mode was stopped and the integrity of the membrane was restored.
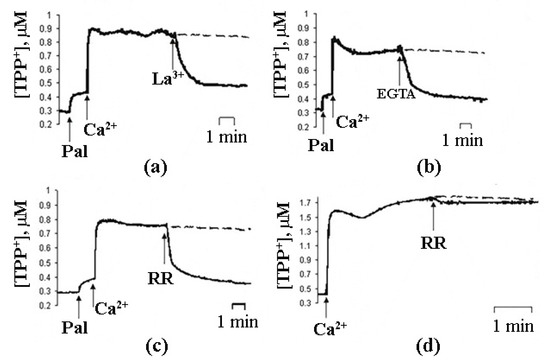
Figure 3. The effects of 5 µM La3+ (a), 1 mM EGTA (b), and 1 µM ruthenium red (RR) (c,d) on the changes in ψ induced by the opening of PA-mPT (a–c) and mPT (d). The incubation medium constable 210 mM mannitol, 70 mM sucrose, 5 mM succinate, 5 µM EGTA, 1 µM rotenone, and 10 mM Hepes/KOH (pH 7.4). Additions in (a–c): Pal (30 nmol/mg protein), Ca2+ (70 nmol/mg protein) and 1 µM CsA; in (d): Ca2+ (200 nmol/mg protein) and 1 mM Pi. Dashed line, control (without RR). Typical traces are shown (n = 7). Adapted from Mironova et al. J. Bioenerg. Biomembr. 39 (2007): 167–174, doi:10.1007/s10863-007-9079-9.
PA/Ca2+-induced membrane permeabilization was also observed in the experiments with SRB-loaded erythrocytes. As can be seen in Figure 1d, the addition of PA did not result in a significant release of SRB from the cells. However, the subsequent addition of Ca2+ triggered a rapid release of the dye [20].
The observations of PA/Ca2+-induced membrane permeabilization on various membrane systems, model and biological, raise the question: are the molecular mechanisms of permeabilization the same in all these cases?
To address this question, we compared characteristics of PA/Ca2+-induced permeabilization of artificial and biological membranes—and it turned out that there were some similarities. Permeabilization of both liposomal and mitochondrial membranes was demonstrated to be inhibited by Ca2+-binding agents (EGTA and bovine serum albumin (BSA)) [18][21], as well as by cholesterol, a modulator of phase state properties of lipid bilayers [22]. Furthermore, liposomes enriched with the mitochondrial lipid cardiolipin were more easily permeabilized, and neutralizing cardiolipin in mitochondria with nonyl acridine orange inhibited permeability transition in the organelles [23][24]. These data suggest that the nature of membrane permeabilization is probably similar in the two types of membranes. They also indicate that the formation of the PA/Ca2+ complexes is a key event in the mechanism of PA-mPT.
As stated above, we hypothesized that PA-mPT was caused by the formation of complexes between PA and Ca2+. The ability to bind Ca2+ with high affinity is a unique feature of long-chain saturated fatty acids (PA and SA (stearic acid)); other lipids and fats have much lower affinities to Ca2+ (Table 1) and cannot induce Ca2+-dependent permeabilization of model and biological membranes [16][19][20][21].
We believe that permeabilization of membranes upon binding of Ca2+ to the anions of long-chain saturated fatty acids is related to a phase transition in the lipid bilayer (Figure 4). This interpretation was supported by the experiments with nonyl acridine orange (NAO), a self-quenching fluorescent probe which can be used to detect changes in the area of liquid phase of the lipid bilayer. Incorporation of PA molecules into liposomal membranes caused their expansion and a corresponding increase in the fluorescence intensity of NAO. The subsequent addition of Ca2+ decreased NAO fluorescence, suggesting a segregation of PA/Ca2+ complexes into solid-phase membrane domains [15].

Figure 4. Schematic representation of hydrophilic lipid pore formation in the liposome membrane during a chemotropic phase transition. Adapted from Agafonov et al. J. Membrane Biol. 215 (2007): 57–68, doi:10.1007/s00232-007-9005-4.
The phase separation induced by Ca2+ in PA-containing membranes can be accompanied by the formation of pores in the lipid bilayer. The mechanism of pore formation under such “chemotropic” phase transition was described earlier by Antonov et al. [25][26]. In respect to PA and Ca2+, the mechanism can be described as follows. The binding of Ca2+ to PA anions at one side of a membrane will cause segregation of PA/Ca2+ complexes into solid-phase domains (Figure 4). Since solid-phase domains are more tightly packed, the overall area of the monolayer will be reduced. Thus, the monolayer containing solid-phase domains will become expanded, and the opposing monolayer will be condensed (stage 2; Figure 4, middle panel). At a certain point, the expanded monolayer may break, and the hydrophobic parts of the bilayer will be exposed to the aqueous phase, producing a highly unstable structure. Consequently, the condensed monolayer would also break and then fuse with the expanded monolayer, closing the hydrophobic edges of the rupture. The resulting structure will be a hydrophilic lipid pore (stage 3; Figure 4, right panel) [27][28][29].
Formation of hydrophilic lipid pores under the conditions when the bilayer is destabilized or stretched is a well-known phenomenon: the pores can be formed, for example, in the process of temperature-induced phase transition, as a result of osmotic shock, or can be produced by the electroporation technique [30][31]. One of the most important properties of lipid pores is their ability to spontaneously close after a short period of time (fractions of a second). In the case of PA and Ca2+, the diameter of such transient pores is approximately 4 nm, which is slightly bigger than the diameter of the classical mPTP, whose size is believed to be in the rage of 2–3 nm [16][19]. Moreover, unlike lipid pores, mPTP is believed to be a channel which can remain open for quite a long time. Furthermore, the formation of PA-mPT pores can be inhibited by RR, which does not affect the probability of mPTP opening [17][18]. The fact that mitochondria can keep operating for a while after PA-mPT is triggered may be, in our opinion, a key feature which enables regulation of many physiological and pathological processes, including apoptosis.
When mitochondria are incubated in the presence of PA, the addition of Ca2+ causes permeability transition that is not inhibited by CSA (Figure 1c) [17][19]. These observations might appear in contrast to the previous reports that PA can potentiate the opening of the classical CSA-sensitive pore. Traditionally, the stimulatory effect of PA on mPTP was explained by either interaction of PA with the protein complexes of the inner mitochondrial membrane or its protonophore activity [32]. However, all those experiments were conducted in the presence of other mPTP activators (namely, orthophosphate and oxidants), which makes it difficult to estimate the specific contribution of PA to the process.
PA-mPT demonstrates properties that distinguish it from the CSA-dependent mPT. First of all, the formation of PA-mPT pores requires much lower concentrations of Ca2+ compared to the classical mPT [13][19][33]. Albeit higher than the basal level of intracellular Ca2+, these concentrations are within the range of the pathological concentrations of intracellular Ca2+ observed during stress [34].
In our experiments, PA/Ca2+-dependent mitochondrial swelling was induced when PA was added at the amount of 30 nmol/mg protein [21]. The most common free fatty acids of the inner mitochondrial membrane are palmitic, stearic, and oleic (6.61, 6.46, and 5.06 molar % of the total lipids, respectively). The combined content of long-chain saturated free fatty acids (PA + SA) in the intact mitochondrial membrane is, therefore, lower than the content necessary to trigger permeability transition (it is estimated to be about 17–18 molar %). Under stress conditions, however, the content of free fatty acids in the mitochondrial membrane can be increased.
The increase in the content of free fatty acids in cellular membranes under stress and pathological conditions is known to be related to the elevation of the Ca2+ concentration inside the cell. Ca2+ activates Ca2+-dependent phospholipases, including mitochondrial phospholipase A2 (PLA2) [35]. PLA2 mostly hydrolyzes phospholipids at the 2nd carbon atom of glycerol, which is primarily esterified by arachidonic acid—yet the enzyme is also capable of cleaving lipid molecules at the 1st position, which is often occupied by saturated fatty acids [33][36]. Correspondingly, the activation of PLA2 by Ca2+ and/or oxidative stress leads to the release of various fatty acids, including PA [36][37][38].
There are some interesting facts about the cell lines that have increased contents of PA and SA (in both free and esterified forms). Specifically, WEHI-164 cells, which are highly prone to apoptotic cell death, are found to have an increased content of PA and SA as compared to the apoptosis-resistant C6 cells [17]. We have also found that, unlike common Wistar rats, the stress-resistant animals of the August line are much more susceptible to the formation of PA-mPT pores (particularly, at Ca2+ concentrations of 2.5–10 µM) [39].
PA-mPT and mPT seem to have different molecular mechanisms—and their properties, as well as regulation, also differ.
As stated above, PA-mPT is not inhibited by CSA, a well-known inhibitor of mPT. Similarly, some other mPT modulators, including inhibitors Adenosine di-phosphate (ADP) and 2,4-Dinitrophenol (DNP) and activators Atractyloside (ATR), orthophosphate, and temerasol, do not affect the probability of PA-mPT [19].
Furthermore, unsaturated fatty acids, which are known to facilitate mPT, cannot induce the emergence of PA-mPT pores. Right the opposite, their presence in the mitochondrial membrane reduces the probability of PA-mPT [24][33].
Another condition necessary for the emergence of PA-mPT pores is the accumulation of Ca2+ in the mitochondrial matrix. Prevention of Ca2+ uptake by mitochondria—by the blockage of Mitochondrial Calcium Uniporter (MCU) with RR or by membrane depolarization—suppresses PA-mPT [18][19]. Thus, membrane potential is necessary for the emergence of PA-mPT pores, whereas mPTP opening can be triggered by membrane depolarization. Additionally, PA-mPT can be induced not only by Ca2+, but also by Sr2+ and, to a lesser extent, by Ba2+ [29]. All these ions can be transported into the mitochondrial matrix by MCU. This also sets PA-mPT apart from mPT, which can only be triggered by Ca2+.
One of the most important properties of lipid pores is their ability to close spontaneously if their diameter does not exceed a certain critical value. For a liquid bilayer phase, this value was estimated to be ~9 nm. The closure of lipid pores occurs rapidly (they exist for a fraction of a second) and restores the integrity of the lipid bilayer [26]. PA-mPT pores are an example of such transient pores. The size of PA-mPT pores is estimated to be ~4 nm, which is slightly larger than the size of mPTP (~2–3 nm). The rapid closure of PA/Ca2+-induced lipid pores was demonstrated in the experiments with liposomes. On the other hand, rapid closure is not something usually observed in the case of mPTP, which often remains open for an extended period [16][19]. We suggest that the ability of PA-mPT pores to close spontaneously and rapidly underlies the so-called “flickering” mode of mitochondrial permeability transition, which is reversible and seems to play an important role in the regulation of many physiological and pathological processes, such as the emergency release of mitochondrial Ca2+ or induction of apoptosis [40].
Based on our experimental data, the putative mechanism of PA-mPT pore formation involves several steps (Figure 5). First, elevation of the intracellular concentration of Ca2+ leads to its enhanced uptake by mitochondria. Second, the accumulation of Ca2+ in the mitochondrial matrix results in the activation of PLA2 and raises the content of free fatty acids in the mitochondrial inner membrane. Third, the anions of long-chain saturated fatty acids form complexes with Ca2+ on the matrix side of the inner mitochondrial membrane and when their content reaches a certain threshold, they segregate into solid domains, which is accompanied by the formation of lipid pores.
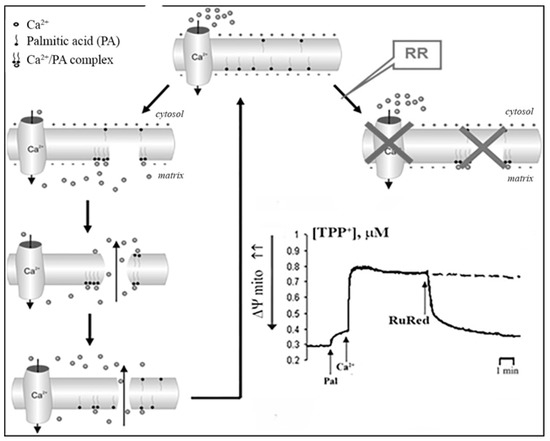
Figure 5. A possible mechanism of PA-mPT pore formation upon the activation of phospholipase A2.
The formation of pores leads to the mitochondrial depolarization and redistribution of ions across the mitochondrial membrane (in particular, the release of Ca2+ and reuptake of H+ by the organelles). Following the rapid closure of pores, membrane potential quickly recovers, owing to enhanced operation of the respiratory chain. Membrane repolarization leads to the reactivation of Ca2+ uptake and induction of a new uptake/release cycle.
The possibility of such a cycle was demonstrated in the experiments with inhibitors of Ca2+-dependent PLA2 on a model of reversible Sr2+ uptake/release by mitochondria (Figure 6). A stepwise addition of Sr2+ to the suspension of energized rat liver mitochondria in a hypotonic medium (hypotonia was required for the activation of PLA2) was shown to cause a nonselective and reversible increase in the membrane permeability to Sr2+, H+, and K+ [41][42][43]. The addition of Sr2+ also led to a reversible membrane depolarization and CSA-insensitive mitochondrial swelling. We hypothesized that the observed changes were associated with the emergence of non-selective pores, following the formation of complexes of PA and SA with Sr2+ [44]. All these effects were prevented by aristolochic acid, an inhibitor of PLA2, further supporting the idea that the free fatty acids released by PLA2 are involved in the permeability transition. Notably, the addition of PA reversed the inhibitory effect of aristolochic acid and restored ion cycling across the membrane (Figure 6), emphasizing the key role of long-chain saturated fatty acids in the mechanism of membrane permeabilization.
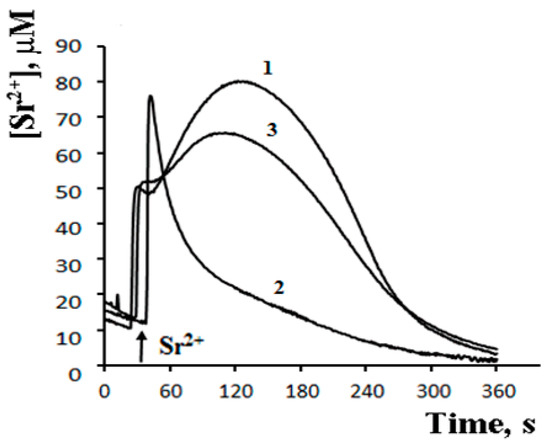
Figure 6. Palmitic acid re-induces the Sr2+-induced permeability transition suppressed by the Ca2+-dependent phospholipase A2 inhibitor—AACOCF3. The medium contained 20 mM sucrose, 1 mM KCl, 1 μM TPP+, 1 μM CsA, 5 mM succinic acid/Tris (pH 7.3). Addition: 40 nmol SrCl2/mg of protein. 1—control; 2—mitochondria preincubated with 15 μM AACOCF3; 3—mitochondria preincubated with 15 μM AACOCF3 and 40 μM PA. The concentration of the mitochondrial protein was 2 mg/mL. Typical curves are shown (n = 4). Adapted from Mironova et al. Biochim. Biophys. Acta. 1848 (2015): 488–495.
As can be seen in Figure 7, after the completion of a Sr2+ uptake/release cycle, mitochondria are functionally active, and the cycle can be repeated again in response to another Sr2+ pulse.
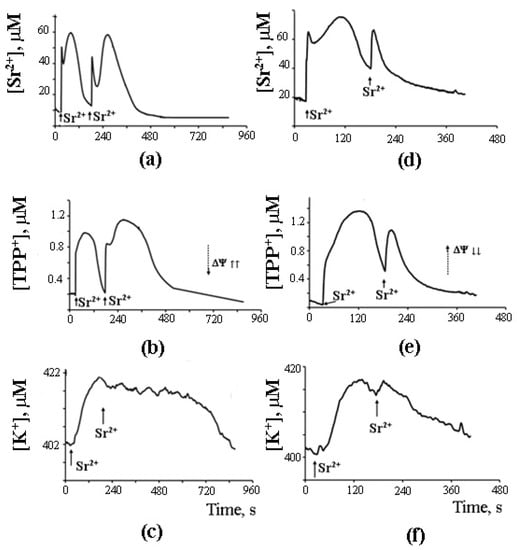
Figure 7. Sr2+-induced cyclic changes in the external concentrations of Sr2+, TPP+, and K+ ions in the suspension of rat liver mitochondria in the absence (a–c) or presence (d–f) of 15 μM AACOCF3 (phospholipase A2 inhibitor). Addition: 47 nmol SrCl2/mg of the protein. The medium was the same as in Figure 6. The concentration of the mitochondrial protein was 2 mg/mL. Typical curves are shown (n = 6). Adapted from Mironova et al. Biochim. Biophys. Acta. 1848 (2015): 488–495.
What is important, though, is that the second cycle cannot be triggered before the first cycle has been completed. Apparently, there is some kind of “refractory” period, during which mitochondria are restoring their functional state [44]. A similar observation was made earlier by Holmuhamedov et al. [43].
On the basis of the experimental data obtained, we propose that PA-mPT pores play a protective role, releasing excessive Ca2+ from mitochondria—and thus can be called “pores of life.” As outlined in the previous section, a pulse of Sr2+ added to mitochondria in the presence of CSA triggers prolonged oscillations of functional parameters of the organelles: K+ and Sr2+ fluxes, membrane potential, pH, matrix volume, rate of oxygen consumption, and H2O2 production. These oscillations can be blocked by the inhibitors of Ca2+-dependent PLA2 (Figure 7). The fact that the oscillatory mode can be maintained for quite a long time suggests a possibility of similar oscillatory processes occurring in vivo, where they could be synchronized with the oscillations of cellular Ca2+. It is also possible that the formation of PA-mPT pores in such a “flickering” mode protects mitochondria from the pathological Ca2+ overload and prevents the permanent opening of CSA-sensitive mPTP.
Thus, PA-mPT forms a system of rapid Ca2+ release and H+ uptake, which is triggered in emergency situations accompanied by Ca2+ overload of mitochondria. It is also possible that the system may operate, to some extent, under normal circumstances, serving the same purpose as K+/H+ and Ca2+/H+ exchangers: maintaining ion homeostasis in the cell.
The ideas that fatty acids can be involved in the release of Ca2+ from mitochondria and that this process is regulated by PLA2 were proposed over 40 years ago [45][46][47][48]. We continued developing these ideas in our works, considering the mechanism of membrane permeabilization in more detail and from a different perspective. We also obtained new evidence of the physiological role of the mechanism—in particular, observations that the susceptibility of mitochondria to the formation of PA-mPT pores is tissue-dependent and changes with age [49]. Another interesting observation was made towards hypoxia-resistant rats: their mitochondria turned out to be more susceptible to the permeabilizing effect of PA and Ca2+ as compared to the organelles of the control animals [50]. In other words, the resistance of animals to hypoxia seems to be associated with the ability of their mitochondria to form PA-mPT pores. This supposition was also supported by the results of measurements of H2O2 production and membrane potential.
It should be noted that the classical mPTP was also proposed to have a flickering mode of operation and thus plays a protective role [51]. Indeed, a transient opening of mPTP should be sufficient to induce Ca2+ release from mitochondria followed by the restoration of their normal function. On the other hand, the lipid pore mechanism might be advantageous in this respect to the protein pore mechanism. First, lipid pores close spontaneously, which ensures that the membrane loses its integrity only for a short time. Further, the involvement of PLA2 guarantees that the lipid pore mechanism is strictly Ca2+-dependent [52][53]. Finally, a high rate of phosphorylation favors the formation of PA-mPT pores [39][50], which makes the lipid pore mechanism physiologically relevant. Overall, the involvement of mPT and PA-mPT in maintaining mitochondrial and cellular Ca2+ homeostasis requires further investigation.





