 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jinwei Zhang | + 6497 word(s) | 6497 | 2021-02-07 05:09:40 | | | |
| 2 | Rita Xu | -3440 word(s) | 3057 | 2021-02-07 10:17:44 | | |
Video Upload Options
Stroke is one of the major culprits responsible for morbidity and mortality worldwide, and the currently available pharmacological strategies to combat this global disease are scanty. Cation-chloride cotransporters (CCCs) are expressed in several tissues (including neurons) and extensively contribute to the maintenance of numerous physiological functions including chloride homeostasis.
1. Introduction of Cation-Chloride Cotransporter Family
The family of cation-chloride cotransporters (CCCs) comprises the Na+-K+-Cl−, Na+-Cl−, and K+-Cl− cotransporters (NKCCs, NCC, and KCCs). Identification of these CCCs in several tissues such as red blood cells, epithelia, and neurons have alluded to their extensive contributions to ion and water homeostasis, both at a cellular and trans-epithelial level [1][2][3]. The identification of the functional properties of most of these transporters dates back to the late 1970s and early 1980s as Cl−-dependent cation fluxes, with red blood cells and Ehrlich ascites tumor cells constituting pivotal model tissues [4][5][6][7]. Subsequently, their molecular identities were established about a decade afterwards [8][9][10]. CCCs are intrinsic membrane proteins that move Na+, K+, and Cl- ions across plasma membranes in a tightly coupled electroneutral manner. They facilitate secondary active transport driven by the gradients generated by the Na+/K+-ATPase [11]. The solute carrier family 12 (SLC12) of the CCC family consists of nine members [2]. A group of three Na+-dependent inward cotransporters comprises of one Na+-Cl- cotransporter (NCC)—its sole isoform is found in the kidney and encoded by SLC12A3 [9] and two Na+-K+-Cl- cotransporters isoforms (NKCC1 and 2)—NKCC1 is ubiquitous whilst NKCC2 is specifically expressed in the kidney and are encoded by SLC12A2 and SLC12A1, respectively [2]. Na+-independent outward transport of K+ and Cl- is facilitated by four K+-Cl- cotransporters with distinct functional properties (KCC1 [12], KCC2 [13], KCC3, and KCC4 [14][15]). The KCC isoforms are encoded by SLC12A4–7 respectively, of which SLC12A5 (KCC2) is found exclusively in neurons [2]. The additional SLC12 family members, CCC9 and CCC-interacting protein (CIP), are encoded by SLC12A8 and SLC12A9, respectively, and have no physiological role ascribed to them yet [16], though recent genome-wide association studies found novel SLC12A8 variants may be associated with dyslipidemia [17], and SLC12A9 may be involved in feather pecking and aggressive behavior [18] (see Table 1).
All proteins in the CCC family have common functional characteristics. These include (1) the coupled transport of one cation (Na+ and/or K+) per individually transported anion, hence the appellation of electroneutral cotransporters, (2) chloride is always the transported anion, (3) all cotransporters are modulated by variations in cell volume, (4) changes in the intracellular chloride concentration ([Cl-]i) influence the modulation of their expression, and (5) the regulation of CCCs activity is achieved through phosphorylation and dephosphorylation processes [19]. The functional and structural characteristics of the Na+ dependent and Na+ independent branches clearly distinguish the two. The degree of identity amongst the Na+ dependent transporters and the Na+ independent are 50% and 70% respectively. Between the two NKCC isoforms, the degree of identity is 25% [20].
Stroke is one of the major culprits responsible for global death and disability [21]. Currently, there is a paucity of pharmacological strategies to reduce the mental damage as well as the burden triggered by this pathology. Ischemic stroke is the most common type of stroke, which accounts for approximately 85% of the cases of the pathology [22]. Ischemia is the disruption of blood flow and the subsequent depletion of oxygen and glucose. As neuronal components strictly function on aerobic metabolism [23], an ischemia in the brain leads to reduced available ATP levels and ionic imbalance across the neuronal cell membrane [24], which causes irreversible neuronal death, also known as ischemic stroke [25][26][27]. During this process, an imbalance of excitatory glutamate and inhibitory gamma amino acid butyric acid (GABA) further accelerate neuronal demise [28][29][30], subsequently leading to the onset of post-stroke seizures [20][31][32]. Neuronal cells excitation is opposed by inhibitory GABA through the activation of GABAA receptors. The activation of the GABAA receptors is dependent on the chloride transmembrane gradient [23]. Notably, CCCs are the primary regulators of chloride homeostasis in the brain [2][33]. This role is accomplished through the extrusion of Cl- via KCC2 and entry of Cl- via NKCC1 which regulates [Cl-]i. GABA is inhibitory as a result of lower [Cl-]I, driven by higher expression of KCC2. Interestingly, immature neurons express less KCC2 and more NKCC1 leading to a higher [Cl-]i and excitatory GABA. The switch from excitatory to inhibitory during neurodevelopment, a process termed excitatory-to-inhibitory GABA switch, is generated through reduction in NKCC1 level and increase in KCC2 level [34]. The expanding work on CCC influence on neuronal excitability in physiological conditions especially during development and pathological conditions suggest that they could be a new treatment approach for stroke [27].
In view of this, constant updates on the role of CCCs in stroke and their regulation is highly germane for the development of therapeutic drugs in the management of this pathology. Thus, the aim of this review is to summarize the current understanding of functional regulation of the CCCs and particularly the role of NKCC1 and KCC3 cotransporters in the pathogenesis of stroke. Then, the regulatory role of the with-no-lysine kinase (WNKs) family and STE20/SPS1-related proline/alanine rich kinase (SPAK) or oxidative stress response kinase (OSR1) (WNK-SPAK/OSR1) signaling pathway in stroke will be considered. Lastly, current pharmacological treatments for stroke with respect to potent inhibitors of WNK-SPAK/OSR1 pathway and NKCC1 cotransporter, and activators of KCC3 transporter will be discussed in this review.
Table 1. The solute carrier family 12 (SLC12) of cation-chloride cotransporters in neurological disorders and others. TAL: thick ascending loop of Henle; DCT: distal convoluted tubule; RVI: regulatory volume increase; RVD: regulatory volume decrease; ND: no data (or none). Functional regulation of the cation-chloride cotransporter family.
|
Encoding Gene |
Co-Transport Ions |
Tissue Distribution |
Physiological Functions |
Genetic Disorders |
References |
|
SLC12A1 (NKCC2) |
Na+, K+, Cl− |
Kidney-specific (TAL) |
NaCl reabsorption in the TAL; regulation of Ca2+ excretion; urine concentration |
Bartter’s syndrome |
|
|
SLC12A2 (NKCC1) |
Na+, K+, Cl− |
Ubiquitous |
Cell volume regulation (RVI); provide ions for secretion |
Potential role in human schizophrenia multi-organ system failure, congenital hydrocephalus, hearing, and neurodevelopmental disorder |
|
|
SLC12A3 (NCC) |
Na+, Cl− |
Kidney-specific (DCT) |
NaCl reabsorption in the DCT; regulation of Ca2+ and K+ renal excretion; |
Gitelman’s syndrome |
|
|
SLC12A4 (KCC1) |
K+, Cl− |
Ubiquitous |
cell volume regulation (RVD), KCl epithelial Transport |
ND |
|
|
SLC12A5 (KCC2) |
K+, Cl− |
Neuron-specific |
Intraneuronal Cl- Concentration regulation |
Idiopathic generalized epilepsy, developmental apoptosis, neurodevelopmental pathology, Rett syndrome |
|
|
SLC12A6 (KCC3) |
K+, Cl− |
Widespread |
Volume regulation in the brain; K+ recycling in the kidney |
Anderman’s syndrome, Charcot–Marie–Tooth disease, hydrocephalus, sensorimotor neuropathy |
|
|
SLC12A7 (KCC4) |
K+, Cl− |
Widespread |
Participates in acid excretion in alpha intercalated cells of collecting duct |
ND |
|
|
SLC12A8 (CCC9) |
Unknown |
Widespread |
No function ascribed yet |
Psoriasis, dyslipidemia |
|
|
SLC12A9 (CIP) |
Unknown |
Widespread |
No function ascribed yet |
May be involved in feather pecking and aggressive behavior |
Undoubtedly, for a cell to function properly it is essential to maintain constant intracellular ionic milieu [53]. Homeostasis of [Cl−]i in particular, influences the movement of fluid across epithelia, the polarity of GABA, and more. The electroneutral CCCs are critical determinants of [Cl−]i [53][60]. The [Cl−]i gradient across the neuronal membrane is crucial for controlling the polarity of GABAergic signaling. GABAA conducts Cl− ions. The direction of Cl− movement through GABAA, which determines whether it is excitatory or inhibitory, is dependent on the [Cl−]i gradient. Entry of Cl− through GABAA results in the hyperpolarization of neurons and the extrusions of Cl- through GABAA depolarizes the neurons [23][61][62][63]. Changes in expression levels of the CCCs during development reverses the chloride gradient in neurons, generating a switch from an excitatory GABA to an inhibitory GABA [23][64]. NKCCs facilitate Cl− movement into the cell, while KCCs facilitates Cl− movement out of the cell. Thus, NKCCs promote an increased participation of [Cl−]i in the pathways for regulatory volume increase (RVI), while the KCCs promote decrease in the [Cl−]i as one of the regulatory volume decrease (RVD) mechanisms [19][53][65] (Figure 1). These evolutionarily conserved transporters are amongst the most important mediators of ion transport in multicellular organisms, with particular importance in mammalian central nervous system (CNS) regulation of ionic and water homeostasis [66]. As mentioned earlier, the CCCs are involved in several important cellular functions such as trans-epithelial ion transport, cell volume regulation, and maintenance of [Cl−]i. Their importance in physiological function is evident by the many human Mendelian disorders of the brain and renal phenotype that arise due to mutations in some members of the CCC family and their upstream regulators [19] (Table 1). For instance, reduction in neuronal KCC2 activity results in decreased inhibition and a hyper-excitable network, a feature shared amongst numerous neurological disorders including epilepsy, autism, post-surgical complication, neuropathic pain, and neuropsychiatric disorders [48][53][67][68].
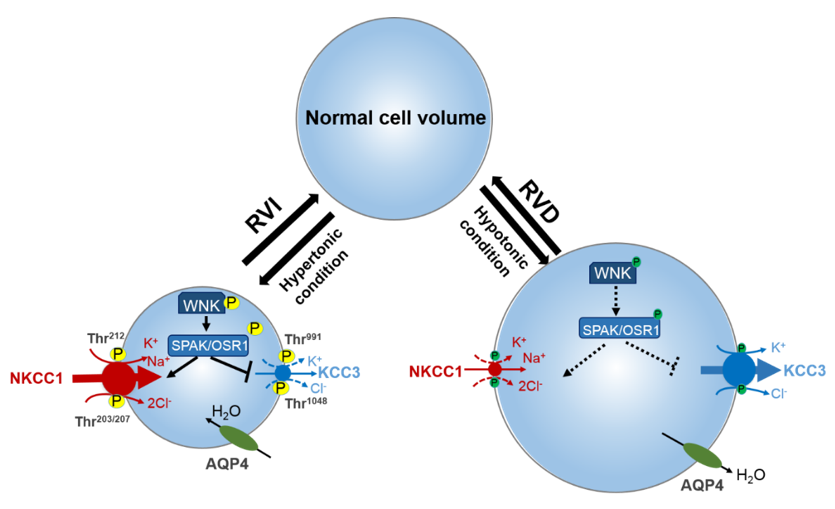
Figure 1. Roles of cation-chloride cotransporters (CCCs) in cell osmoregulation. Intracellular osmolarity changes activate cellular volume regulation. Under hypertonic extracellular conditions of cell shrinkage due to water extrusion from the cell, a counter-response of regulatory volume increase (RVI) restores normal cell volume. In this condition, the WNK-SPAK/OSR1 pathway is activated leading to the phosphorylation of the CCCs. This activates NKCC1 and inhibits KCC, leading to the NKCC1-mediated influx of Na+, K+, and Cl− along with water, thus restoring cell volume. On the contrary, under hypotonic stress conditions of cell swelling, the cell activates a regulatory volume decrease (RVD). The WNK-SPAK/OSR1 pathway remains inactive and NKCC1 and KCCs are dephosphorylated. This stimulates KCC3 but inhibits NKCC1 leading to the efflux K+ and Cl− along with water, and cell volume decrease. NKCC1, K+-Cl− cotransporters; KCC3, K+-Cl− cotransporter 3; WNK, with-no-lysine kinase; SPAK, STE20/SPS1-related proline/alanine rich kinase; OSR1, oxidative stress response kinase; AQP4, aquaporin. Part of figure elements were adapted from Huang et al. [65].
Since CCCs are key players in several important cellular functions and principally responsible for reciprocal cations (Na+ and K+) exchange with Cl- to maintain cellular balance, regulatory mechanisms are crucial to coordinate their activity [53][60][67][69]. Indeed, several kinases and phosphatases regulate their transport activity. However, previous reports have established that members of the WNKs family and their downstream targets, SPAK and OSR1, are the master regulators of CCCs activity [1][53][60][65][70][71][72][73][74][75].
Though it has been appreciated for some three decades that protein phosphorylation coupled with external osmotic environment are crucial components in regulation of CCC activities [2][54][76][77][78][79][80], knowledge on the enzymes that regulate these signaling networks was sparse then. The WNK family encoded by the genes WNK1–4 [81], SPAK, and OSR1 play crucial roles in the regulation of cell volume homeostasis through the regulation of intracellular Na+, K+, and Cl- [53][82]. The many roles of the WNK-SPAK/OSR1-CCC pathway which include cell volume homeostasis, epithelial transport, and GABA signaling are associated with an array of pathologies which include essential hypertension, cerebral edema, anemia, and neuropathic pain [1][19][53][60][65][67][71][73]. In response to osmotic stress of low [Cl−]i, isoforms of WNK are activated through phosphorylation. The WNK isoforms then phosphorylate the related downstream kinases SPAK and/or OSR1 [83][84]. Activated SPAK and/or OSR1 phosphorylates the CCCs, which activates NCC, NKCC1, and NKCC2 but inhibits KCCs through a reciprocal regulatory mechanism [67][72] (Figure 2). The counter regulation of the CCCs coordinates Cl- movement across the membrane to maintain Cl- homeostasis and circumvent superfluous energy utilization [65].
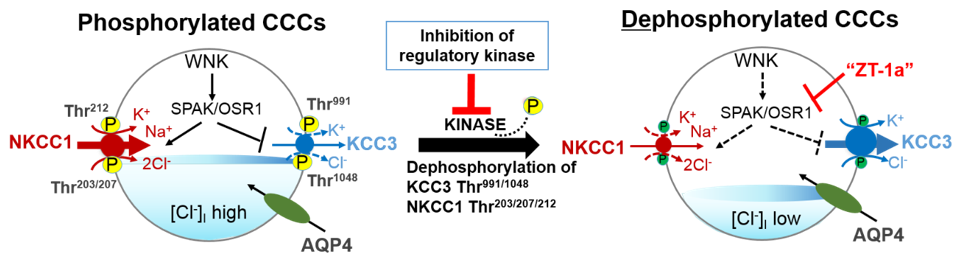
Figure 2. A novel strategy to facilitate cellular Cl− extrusion by coincident NKCC1 inhibition and KCC3 activation by inhibiting Table 1 kinases. Reversible serine-threonine phosphorylation reciprocally regulates NKCC1 and KCC3. Hypotonic low [Cl−]i conditions or a reduction in cell volume activates the WNK-SPAK/OSR1 pathway to promote Cl− and water influx. This leads to the phosphorylation of NKCC1 and KCC3 and their activation and inhibition respectively. When [Cl−]i becomes too high or cell volume increases, WNK-SPAK/OSR1 pathway is inhibited. The cotransporters are dephosphorylated, KCC3 is activated and facilitates [Cl−]i and water efflux to restore ion and osmotic homeostasis. CCCs, cation-chloride cotransporters; NKCC1, K+-Cl− cotransporters; KCC3, K+-Cl− cotransporter 3; WNK, with-no-lysine kinase; SPAK, STE20/SPS1-related proline/alanine rich kinase; OSR1, oxidative stress response kinase; AQP4, aquaporin 4; ZT-1a, specific SPAK inhibitor. Part of figure elements were adapted from Salihu et al. [85].
To maintain cell volume homeostasis, the WNK-SPAK/OSR1 kinase pathway activates NKCC1 and simultaneously inhibits KCCs through phosphorylation. Conversely, dephosphorylation inhibits NKCC1 and activates the KCCs [86]. The major phosphorylation sites of NKCC1 include Thr203, Thr207, and Thr212 in the N-terminus whilst the phosphorylation sites of KCC1–4 are located in the C-terminus (Thr991 and Thr1048 in KCC3 and Thr906 and Thr1007 in KCC2) [59][72]. Notably, the phosphorylation sites on KCC3, Thr991 and Thr1048, are conserved amongst all KCC isoforms in humans [72]. Substitution of these threonine residues that make up the sites of regulated phosphorylation inhibited the phosphorylation and subsequent activation of KCC2 and KCC3 [86][87][88]. Recently, it was established that the WNK3-SPAK complex is critical for regulated phosphorylation of KCC3 Thr991 and Thr1048 residues [86] (also see Figures 3 and 4).
Specific conserved carboxyl-terminal (CCT) domains on SPAK/OSR1 interact with NKCC1 and KCCs [80][86][89]. The Arg-Phe-Xaa-Val/Ile (RFXV/I) domain located in the N-terminal of NKCCs and KCCs is able to recognize the SPAK CCT domain (Figures 3 and 4). Interestingly, a subtype of the KCC2 isoform, KCC2b lacks the RFXV/I motif to facilitate interaction with SPAK. Thus, only KCC2a transport activity decreased when SPAK was overexpressed [53][90]. This interaction of SPAK/OSR1 with both the upstream WNKs and downstream CCCs is crucial for coordinating CCC cellular activity in various osmotic conditions [89][91]. The binding of WNK to SPAK/OSR1 allows for the phosphorylation of residues in the T-loop of the SPAK catalytic domain required for SPAK activation [89][91]. Only once activated is SPAK then able to phosphorylate and inhibit KCC2 and KCC3 at Thr1048 and Thr1007 respectively and activate NKCC1 at Thr203/Thr207/Thr212. These processes are essential in response to cellular shrinkage and hypertonicity (Figure 1) [53]. In hypertonic conditions, SPAK/OSR1 phosphorylation and activation of NKCC1 is key to achieve RVI [53][67] as an influx of Na+, K+, Cl- through the NKCC1 along with water will allow for cell volume recovery (Figure 1). Under hypotonic extracellular conditions, water enters the cells and causes cell swelling, subsequently triggering a counter-volume regulation response (RVD). The WNK-SPAK/OSR1 pathway in this condition remains inactive and inhibits NKCC1 activity (Figure 1). Furthermore, the dephosphorylation of KCCs mediated by phosphatase stimulates KCC activity and causes efflux of K+ and Cl- along with water, decreasing cell volume (Figure 1) [65]. Thus, pharmacological or genetic antagonistic events of WNK-SPAK/OSR1 will lead to a [Cl-]i efflux coupled through simultaneous dephosphorylation of NKCC1 and KCCs. This will then mitigate energy failure occasioned by osmotic stress, as evident in some neurological disturbances such as cerebral edema [32][65][67][86][92].
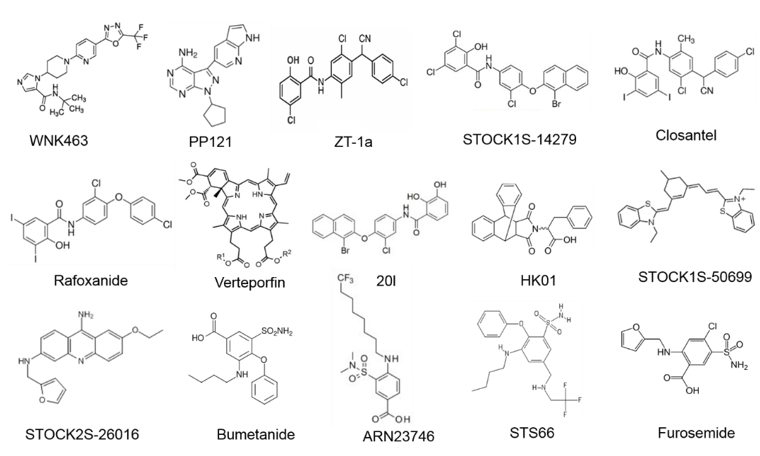
Figure 3. Molecular structures of WNK-SPAK/OSR1-CCCs signaling pathway compounds.
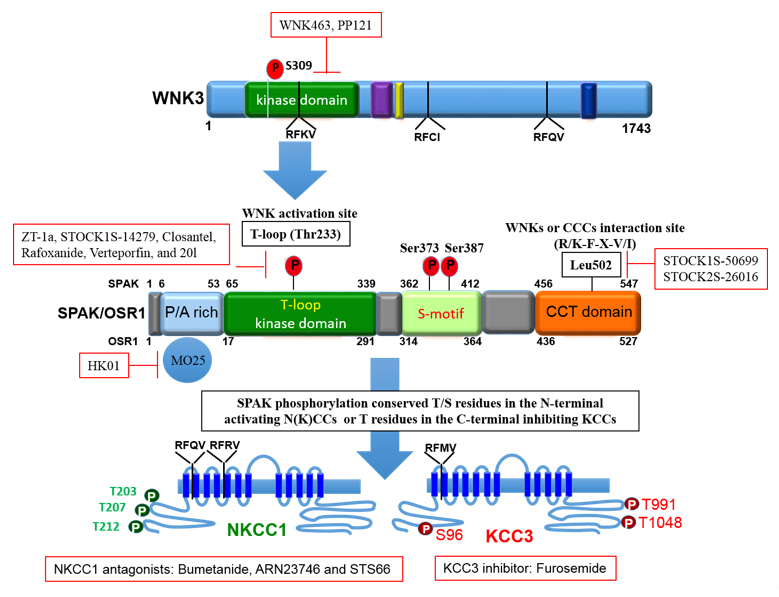
Figure 4. The domain structure of SPAK and the phosphorylation target sites on NKCC1 and KCC3. OSR1 lack the P/A rich (PAPA) domain that is present in SPAK. The figure depicts small molecule inhibitors that target the WNK-SPAK-CCC signaling pathway and their sites of actions. STOCK1S-50699 and STOCK2S-26016 operate through binding to the CCT domain consequently blocking the interaction between SPAK/OSR1 and WNK. STOCK1S-14279, Closantel, Rafoxanide, Verteporfin, and 20l bind the T233E residue on SPAK that is constitutively active or WNK-sensitive. WNK463 and PP121 that inhibit WNKs catalytic activity. HK01, an inhibitor of the mouse protein-25 (M025). Bumetanide, ARN23746 and STS66 are NKCC1 antagonists. Furosemide is a KCC3 inhibitor. ZT-1a is a specific SPAK inhibitor. CCCs, cation-chloride cotransporters; NKCC1, K+-Cl- cotransporters; KCC3, K+-Cl- cotransporter 3; WNK, with-no-lysine kinase; SPAK, STE20/SPS1-related proline/alanine rich kinase; OSR1, oxidative stress response kinase.
2. Role of NNKCC1 in Stroke
NKCCs play crucial roles in regulating neuronal functions. They are abundantly expressed in neurons throughout the brain and are involved in ion homeostasis maintenance and neuronal excitatory functions [93]. Majorly, they function in regulation and repair of nerve injury through GABAergic signaling [1][94]. However, under specific conditions such as cerebral ischemia, the expression of NKCCs can be altered [94]. Overstimulation of NKCC1 and other major glial ion transporters (such as Na+/H+, Na+/Ca2+ and Na+/HCO3- exchangers) can contribute to glial apoptosis, inflammation, demyelination, inflammation, and excitotoxicity [26]. This cascade of events is involved in the development and progression of neurological diseases such as stroke [26]. Studies have demonstrated evidence of increase NKCC1 expression in neurons, a phenotype resembling immature neurons, following an ischemic stroke [95][96][97]. The altered NKCC1 expression observed post stroke may be responsible for the increased in Na+ and Cl– levels in neurons leading to a GABA-mediated depolarization. These events also contribute to a hyper-excitable neuron and cell swelling occasioned by cerebral ischemia [95][98]. Furthermore, disrupted endoplasmic reticulum Ca2+ homeostasis [99] and elevated extracellular levels of potassium, glutamate, interleukin-6 [100], interleukin-18 [101], interleukin 1β, and tumor necrotic factor-α [102] which happen during/post cerebral ischemia have been shown to stimulate NKCC1 mRNA gene expression in both neurons and astrocytes. Notably, the elevated extracellular potassium levels seems to be Ca2+-dependent as NKCC1 activation is completely terminated either through the removal of extracellular calcium or using Nifedipine to block L-type voltage-dependent calcium channels [103]. Comparatively, similar effects were seen in the expression of NKCC1 mRNA gene in white and gray matter of mutant and wild-type (WT) mice [104]. In addition, [105] an epigenetic study using quantitative real-time RT-PCR technique on cortical slice culture from rats suggested that DNA methylation/demethylation contribute to the regulation of NKCC1 expression during postnatal development and in response to neuronal injury (ischemia) [105].
Following ischemic stroke, both NKCC1 and KCCs are phosphorylated via the WNK-SPAK/OSR1 signaling pathway, leading to NKCC1 activation and KCC inhibition [22][65]. Other contributors leading to NKCC1 activation following an ischemia include: the WNK-calcium binding protein (Cab39; [106]) as well as antagonists of V1 vasopressin [107], MAPK (p38, ERK, JNK, Raf) pathways, cAMP response element-binding protein (CREB) phosphorylation and the ubiquitous transcription factor; hypoxia inducible factor 1-alpha (HIF-1α). This leads to the stimulation of vascular endothelial growth factor (VEGF) expression and ultimate onset of ischemic stroke [37][108][109][110]. Studies on the human subacute ischemic stroke brain tissues demonstrate increased NKCC1 mRNA gene expression [106][106]. The contribution of NKCC1 protein activation to ischemic brain havocs is now evident as genetic deletion of NKCC1 or its upstream regulator WNK3 in mouse transient middle cerebral artery (MCA) occlusion models displayed minimal infarction, edema, and white matter damage [32][104]. Another study demonstrated increased NKCC1 activity in the perilesional cortex of rats challenged with focal cerebral ischemia induced by endothelin-1 (ET-1) [111]. Furthermore, inhibition of NKCC1 has been reported to reduce edema, Na+ uptake, and ischemic injury in rats subjected to STZ-induced hyperglycemic ischemic stroke [112].
References
- Lu, D.C.-Y.; Hannemann, A.; Wadud, R.; Rees, D.C.; Brewin, J.N.; Low, P.S.; Gibson, J.S. The role of WNK in modulation of KCl cotransport activity in red cells from normal individuals and patients with sickle cell anaemia. Pflügers Arch. Eur. J. Physiol. 2019, 471, 1539–1549.
- Gamba, G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Rev. 2005, 85, 423–493.
- Meor Azlan, N.; Koeners, M.; Zhang, J. Regulatory control of the Na-Cl co-transporter NCC and its therapeutic potential for hypertension. Acta Pharm. Sin. B 2020, doi:10.1016/j.apsb.2020.09.009.
- Lauf, P.; Theg, B. A chloride dependent K+ flux induced by N-ethylmaleimide in genetically low K+ sheep and goat erythrocytes. Biophys. Res. Commun. 1980, 92, 1422–1428.
- Dunham, P.B.; Stewart, G.W.; Ellory, J.C. Chloride-activated passive potassium transport in human erythrocytes. Natl. Acad. Sci. USA 1980, 77, 1711–1715.
- Hoffmann, E.; Sjoholm, C.; Simonsen, L. Anion-Cation Cotransport and Volume Regulation in Ehrlich Ascites Tumor-Cells. J. Physiol. Lond. 1981, 319, P94–P95.
- Gibson, J.S.; Ellory, J.C.; Adragna, N.C.; Lauf, P.K. Pathophysiology of the K+-Cl-cotransporters: Paths to discovery and overview. In Physiology and Pathology of Chloride Transporters and Channels in the Nervous System: From Molecules to Disease; Academic Press: London, UK, 2009; pp. 27–42.
- Xu, J.-C.; Lytle, C.; Zhu, T.T.; Payne, J.A.; Benz, E.; Forbush, B. Molecular cloning and functional expression of the bumetanide-sensitive Na-K-Cl cotransporter. Natl. Acad. Sci. USA 1994, 91, 2201–2205.
- Gamba, G.; Saltzberg, S.N.; Lombardi, M.; Miyanoshita, A.; Lytton, J.; Hediger, M.A.; Brenner, B.M.; Hebert, S.C. Primary structure and functional expression of a cDNA encoding the thiazide-sensitive, electroneutral sodium-chloride cotransporter. Natl. Acad. Sci. USA 1993, 90, 2749–2753.
- Pellegrino, C.M.; Rybicki, A.C.; Musto, S.; Nagel, R.L.; Schwartz, R.S. Molecular identification and expression of erythroid K: Cl cotransporter in human and mouse erythroleukemic cells. Blood Cells Mol. Dis. 1998, 24, 31–40.
- Piechotta, K.; Lu, J.; Delpire, E. Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1). Biol. Chem. 2002, 277, 50812–50819, doi:10.1074/jbc.M208108200.
- Gillen, C.M.; Brill, S.; Payne, J.A.; Forbush, B. Molecular cloning and functional expression of the K-Cl cotransporter from rabbit, rat, and human A new member of the cation-chloride cotransporter family. Biol. Chem. 1996, 271, 16237–16244.
- Payne, J.A. Functional characterization of the neuronal-specific K-Cl cotransporter: Implications for [K+] oregulation. J. Physiol. Cell Physiol. 1997, 273, C1516–C1525.
- Hiki, K.; D’Andrea, R.J.; Furze, J.; Crawford, J.; Woollatt, E.; Sutherland, G.R.; Vadas, M.A.; Gamble, J.R. Cloning, characterization, and chromosomal location of a novel human K+-Cl− J. Biol. Chem. 1999, 274, 10661–10667.
- Mercado, A.; Song, L.; George, A.; Delpire, E.; Mount, D. Molecular, functional, and genomic characterization of KCC3 and KCC4. Am. Soc. Nephrol. 1999, 10, 38A.
- Gagnon, K.B.; Delpire, E. Physiology of SLC12 transporters: Lessons from inherited human genetic mutations and genetically engineered mouse knockouts. J. Physiol. Cell Physiol. 2013, 304, C693–C714.
- Wang, G.; Huang, H.; He, Y.; Ruan, L.; Huang, J. Bumetanide protects focal cerebral ischemia-reperfusion injury in rat. J. Clin. Exp. Pathol. 2014, 7, 1487.
- Wilkinson, C.M.; Fedor, B.A.; Aziz, J.R.; Nadeau, C.A.; Brar, P.S.; Clark, J.J.; Colbourne, F. Failure of bumetanide to improve outcome after intracerebral hemorrhage in rat. PLoS ONE 2019, 14, e0210660.
- de los Heros, P.; Pacheco-Alvarez, D.; Gamba, G. Role of WNK kinases in the modulation of cell volume. In Current Topics in Membranes; Elsevier: Amsterdam, The Netherlands, 2018; Volume 81, pp. 207–235.
- Yang, T.; Zhao, K.; Shu, H.; Chen, X.; Cheng, J.; Li, S.; Zhao, Z.; Kuang, Y.; Yu, S. The Nogo receptor inhibits proliferation, migration and axonal extension by transcriptionally regulating WNK1 in PC12 cells. Neuroreport 2017, 28, 533–539, doi:10.1097/WNR.0000000000000800.
- Johnson, W.; Onuma, O.; Owolabi, M.; Sachdev, S. Stroke: A global response is needed. World Health Organ. 2016, 94, 634.
- Martín-Aragón Baudel, M.A.; Poole, A.V.; Darlison, M.G. Chloride co-transporters as possible therapeutic targets for stroke. Neurochem. 2017, 140, 195–209.
- Schulte, J.T.; Wierenga, C.J.; Bruining, H. Chloride transporters and GABA polarity in developmental, neurological and psychiatric conditions. Biobehav. Rev. 2018, 90, 260–271.
- Mayor, D.; Tymianski, M. Neurotransmitters in the mediation of cerebral ischemic injury. Neuropharmacology 2018, 134, 178–188.
- Boscia, F.; Begum, G.; Pignataro, G.; Sirabella, R.; Cuomo, O.; Casamassa, A.; Sun, D.; Annunziato, L. Glial Na+-dependent ion transporters in pathophysiological conditions. Glia 2016, 64, 1677–1697.
- Song, S.; Luo, L.; Sun, B.; Sun, D. Roles of glial ion transporters in brain diseases. Glia 2020, 68, 472–494.
- Zagrean, A.-M.; Grigoras, I.-F.; Iesanu, M.I.; Ionescu, R.-B.; Chitimus, D.M.; Haret, R.M.; Ianosi, B.; Ceanga, M.; Zagrean, L. Neuronal Transmembrane Chloride Transport Has a Time-Dependent Influence on Survival of Hippocampal Cultures to Oxygen-Glucose Deprivation. Brain Sci. 2019, 9, 360.
- Mele, M.; Costa, R.O.; Duarte, C.B. Alterations in GABAA-receptor trafficking and synaptic dysfunction in brain disorders. Cell. Neurosci. 2019, 13, 77.
- Smith, K.R.; Muir, J.; Rao, Y.; Browarski, M.; Gruenig, M.C.; Sheehan, D.F.; Haucke, V.; Kittler, J.T. Stabilization of GABAA receptors at endocytic zones is mediated by an AP2 binding motif within the GABAA receptor β3 subunit. Neurosci. 2012, 32, 2485–2498.
- Mielke, J.G.; Wang, Y.T. Insulin exerts neuroprotection by counteracting the decrease in cell-surface GABAA receptors following oxygen–glucose deprivation in cultured cortical neurons. Neurochem. 2005, 92, 103–113.
- Fu, C.-Y.; He, X.-Y.; Li, X.-F.; Zhang, X.; Huang, Z.-W.; Li, J.; Chen, M.; Duan, C.-Z. Nefiracetam attenuates pro-inflammatory cytokines and GABA transporter in specific brain regions of rats with post-ischemic seizures. Physiol. Biochem. 2015, 37, 2023–2031.
- Begum, G.; Yuan, H.; Kahle, K.T.; Li, L.; Wang, S.; Shi, Y.; Shmukler, B.E.; Yang, S.S.; Lin, S.H.; Alper, S.L.; et al. Inhibition of WNK3 Kinase Signaling Reduces Brain Damage and Accelerates Neurological Recovery After Stroke. Stroke 2015, 46, 1956–1965, doi:10.1161/STROKEAHA.115.008939.
- Russell, J.M. Sodium-potassium-chloride cotransport. Rev. 2000, 80, 211–276.
- Ben-Ari, Y.; Khalilov, I.; Kahle, K.T.; Cherubini, E. The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. Neuroscientist 2012, 18, 467–486.
- Adachi, M.; Asakura, Y.; SATO, Y.; Tajima, T.; Nakajima, T.; Yamamoto, T.; Fujieda, K. Novel SLC12A1 (NKCC2) mutations in two families with Bartter syndrome type 1. J. 2007, 54, 1003–1007.
- Konopacka, A.; Qiu, J.; Yao, S.T.; Greenwood, M.P.; Greenwood, M.; Lancaster, T.; Inoue, W.; de Souza Mecawi, A.; Vechiato, F.M.; de Lima, J.B. Osmoregulation requires brain expression of the renal Na-K-2Cl cotransporter NKCC2. Neurosci. 2015, 35, 5144–5155.
- Wallace, B.K.; Jelks, K.A.; O’Donnell, M.E. Ischemia-induced stimulation of cerebral microvascular endothelial cell Na-K-Cl cotransport involves p38 and JNK MAP kinases. J. Physiol. Cell Physiol. 2012, 302, C505–C517.
- Dixon, M.J.; Gazzard, J.; Chaudhry, S.S.; Sampson, N.; Schulte, B.A.; Steel, K.P. Mutation of the Na-K-Cl co-transporter gene Slc12a2 results in deafness in mice. Mol. Genet. 1999, 8, 1579–1584.
- Nezu, A.; Parvin, M.N.; Turner, R.J. A conserved hydrophobic tetrad near the C terminus of the secretory Na+-K+-2Cl- cotransporter (NKCC1) is required for its correct intracellular processing. Biol. Chem. 2009, 284, 6869–6876.
- Orlov, S.N.; Koltsova, S.V.; Kapilevich, L.V.; Gusakova, S.V.; Dulin, N.O. NKCC1 and NKCC2: The pathogenetic role of cation-chloride cotransporters in hypertension. Genes Dis. 2015, 2, 186–196.
- Töllner, K.; Brandt, C.; Töpfer, M.; Brunhofer, G.; Erker, T.; Gabriel, M.; Feit, P.W.; Lindfors, J.; Kaila, K.; Löscher, W. A novel prodrug-based strategy to increase effects of bumetanide in epilepsy. Neurol. 2014, 75, 550–562.
- Walcott, B.P.; Kahle, K.T.; Simard, J.M. Novel treatment targets for cerebral edema. Neurotherapeutics 2012, 9, 65–72.
- Koumangoye, R.; Bastarache, L.; Delpire, E. NKCC1: Newly Found as a Human Disease-Causing Ion Transporter. Function 2021, 2, zqaa028, doi:10.1093/function/zqaa028.
- Blaesse, P.; Airaksinen, M.S.; Rivera, C.; Kaila, K. Cation-chloride cotransporters and neuronal function. Neuron 2009, 61, 820–838.
- Delpire, E.; Mount, D.B. Human and murine phenotypes associated with defects in cation-chloride cotransport. Rev. Physiol. 2002, 64, 803–843.
- Rust, M.B.; Faulhaber, J.; Budack, M.K.; Pfeffer, C.; Maritzen, T.; Didie, M.; Beck, F.X.; Boettger, T.; Schubert, R.; Ehmke, H.; et al. Neurogenic mechanisms contribute to hypertension in mice with disruption of the K-Cl cotransporter KCC3. Res. 2006, 98, 549–556, doi:10.1161/01.RES.0000204449.83861.22.
- Boettger, T.; Hubner, C.A.; Maier, H.; Rust, M.B.; Beck, F.X.; Jentsch, T.J. Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4. Nature 2002, 416, 874–878, doi:10.1038/416874a.
- Kahle, K.T.; Schmouth, J.F.; Lavastre, V.; Latremoliere, A.; Zhang, J.; Andrews, N.; Omura, T.; Laganiere, J.; Rochefort, D.; Hince, P.; et al. Inhibition of the kinase WNK1/HSN2 ameliorates neuropathic pain by restoring GABA inhibition. Signal. 2016, 9, ra32, doi:10.1126/scisignal.aad0163.
- Mavrovic, M.; Uvarov, P.; Delpire, E.; Vutskits, L.; Kaila, K.; Puskarjov, M. Loss of non-canonical KCC2 functions promotes developmental apoptosis of cortical projection neurons. EMBO Rep. 2020, 21, e48880, doi:10.15252/embr.201948880.
- Hinz, L.; Torrella Barrufet, J.; Heine, V.M. KCC2 expression levels are reduced in post mortem brain tissue of Rett syndrome patients. Acta Neuropathol. Commun. 2019, 7, 196, doi:10.1186/s40478-019-0852-x.
- Pisella, L.I.; Gaiarsa, J.L.; Diabira, D.; Zhang, J.; Khalilov, I.; Duan, J.; Kahle, K.T.; Medina, I. Impaired regulation of KCC2 phosphorylation leads to neuronal network dysfunction and neurodevelopmental pathology. Signal. 2019, 12, eaay0300, doi:10.1126/scisignal.aay0300.
- Garneau, A.P.; Marcoux, A.A.; Frenette-Cotton, R.; Mac-Way, F.; Lavoie, J.L.; Isenring, P. Molecular insights into the normal operation, regulation, and multisystemic roles of K(+)-Cl(-) cotransporter 3 (KCC3). J. Physiol. Cell Physiol. 2017, 313, C516–C532, doi:10.1152/ajpcell.00106.2017.
- Shekarabi, M.; Zhang, J.; Khanna, A.R.; Ellison, D.H.; Delpire, E.; Kahle, K.T. WNK Kinase Signaling in Ion Homeostasis and Human Disease. Cell Metab. 2017, 25, 285–299, doi:10.1016/j.cmet.2017.01.007.
- Torchia, J.; Lytle, C.; Pon, D.; Forbush, B.; Sen, A. The Na-K-Cl cotransporter of avian salt gland. Phosphorylation in response to cAMP-dependent and calcium-dependent secretogogues. Biol. Chem. 1992, 267, 25444–25450.
- Al Shibli, N.; Al-Maawali, A.; Elmanzalawy, A.; Al-Nabhani, M.; Koul, R.; Gabr, A.; Al Murshedi, F. A Novel Splice-Site Variant in SLC12A6 Causes Andermann Syndrome without Agenesis of the Corpus Callosum. Pediatr. Genet. 2020, 9, 293–295, doi:10.1055/s-0039-1700975.
- Jin, S.C.; Furey, C.G.; Zeng, X.; Allocco, A.; Nelson-Williams, C.; Dong, W.; Karimy, J.K.; Wang, K.; Ma, S.; Delpire, E.; et al. SLC12A ion transporter mutations in sporadic and familial human congenital hydrocephalus. Genet. Genom. Med. 2019, 7, e892, doi:10.1002/mgg3.892.
- Park, J.; Flores, B.R.; Scherer, K.; Kuepper, H.; Rossi, M.; Rupprich, K.; Rautenberg, M.; Deininger, N.; Weichselbaum, A.; Grimm, A.; et al. De novo variants in SLC12A6 cause sporadic early-onset progressive sensorimotor neuropathy. Med. Genet. 2020, 57, 283–288, doi:10.1136/jmedgenet-2019-106273.
- Flatman, P.W. Cotransporters, WNKs and hypertension: Important leads from the study of monogenetic disorders of blood pressure regulation. Sci. 2007, 112, 203–216.
- Richardson, C.; Alessi, D.R. The regulation of salt transport and blood pressure by the WNK-SPAK/OSR1 signalling pathway. Cell Sci. 2008, 121, 3293–3304, doi:10.1242/jcs.029223.
- Heubl, M.; Zhang, J.; Pressey, J.C.; Al Awabdh, S.; Renner, M.; Gomez-Castro, F.; Moutkine, I.; Eugene, E.; Russeau, M.; Kahle, K.T.; et al. GABAA receptor dependent synaptic inhibition rapidly tunes KCC2 activity via the Cl(-)-sensitive WNK1 kinase. Commun. 2017, 8, 1776, doi:10.1038/s41467-017-01749-0.
- Rivera, C.; Voipio, J.; Payne, J.A.; Ruusuvuori, E.; Lahtinen, H.; Lamsa, K.; Pirvola, U.; Saarma, M.; Kaila, K. The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 1999, 397, 251–255, doi:10.1038/16697.
- Delpire, E. Cation-chloride cotransporters in neuronal communication. Physiology 2000, 15, 309–312.
- Kaila, K.; Price, T.J.; Payne, J.A.; Puskarjov, M.; Voipio, J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Rev. Neurosci. 2014, 15, 637–654.
- Huberfeld, G.; Wittner, L.; Clemenceau, S.; Baulac, M.; Kaila, K.; Miles, R.; Rivera, C. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. Neurosci. 2007, 27, 9866–9873.
- Huang, H.; Song, S.; Banerjee, S.; Jiang, T.; Zhang, J.; Kahle, K.T.; Sun, D.; Zhang, Z. The WNK-SPAK/OSR1 Kinases and the Cation-Chloride Cotransporters as Therapeutic Targets for Neurological Diseases. Aging Dis. 2019, 10, 626–636, doi:10.14336/AD.2018.0928.
- Kahle, K.T.; Khanna, A.R.; Alper, S.L.; Adragna, N.C.; Lauf, P.K.; Sun, D.; Delpire, E. K-Cl cotransporters, cell volume homeostasis, and neurological disease. Trends Mol. Med. 2015, 21, 513–523.
- Alessi, D.R.; Zhang, J.; Khanna, A.; Hochdorfer, T.; Shang, Y.; Kahle, K.T. The WNK-SPAK/OSR1 pathway: Master regulator of cation-chloride cotransporters. Signal. 2014, 7, re3, doi:10.1126/scisignal.2005365.
- Kahle, K.T.; Khanna, A.R.; Duan, J.; Staley, K.J.; Delpire, E.; Poduri, A. The KCC2 Cotransporter and Human Epilepsy: Getting Excited About Inhibition. Neuroscientist 2016, 22, 555–562, doi:10.1177/1073858416645087.
- Hartmann, A.-M.; Nothwang, H.G. Molecular and evolutionary insights into the structural organization of cation chloride cotransporters. Cell. Neurosci. 2015, 8, 470.
- Zhang, J.; Bhuiyan, M.I.H.; Zhang, T.; Karimy, J.K.; Wu, Z.; Fiesler, V.M.; Zhang, J.; Huang, H.; Hasan, M.N.; Skrzypiec, A.E.; et al. Modulation of brain cation-Cl(-) cotransport via the SPAK kinase inhibitor ZT-1a. Commun. 2020, 11, 78, doi:10.1038/s41467-019-13851-6.
- Zhang, J.; Cordshagen, A.; Medina, I.; Nothwang, H.G.; Wisniewski, J.R.; Winklhofer, M.; Hartmann, A.M. Staurosporine and NEM mainly impair WNK-SPAK/OSR1 mediated phosphorylation of KCC2 and NKCC1. PLoS ONE 2020, 15, e0232967, doi:10.1371/journal.pone.0232967.
- de Los Heros, P.; Alessi, D.R.; Gourlay, R.; Campbell, D.G.; Deak, M.; Macartney, T.J.; Kahle, K.T.; Zhang, J. The WNK-regulated SPAK/OSR1 kinases directly phosphorylate and inhibit the K+-Cl- co-transporters. J. 2014, 458, 559–573, doi:10.1042/BJ20131478.
- Brown, A.; Meor Azlan, N.F.; Wu, Z.; Zhang, J. WNK-SPAK/OSR1-NCC kinase signaling pathway as a novel target for the treatment of salt-sensitive hypertension. Acta Pharmacol. Sin. 2020, doi:10.1038/s41401-020-0474-7.
- AlAmri, M.A.; Kadri, H.; Alderwick, L.J.; Jeeves, M.; Mehellou, Y. The Photosensitising Clinical Agent Verteporfin Is an Inhibitor of SPAK and OSR1 Kinases. Chembiochem 2018, 19, 2072–2080, doi:10.1002/cbic.201800272.
- Rinehart, J.; Vazquez, N.; Kahle, K.T.; Hodson, C.A.; Ring, A.M.; Gulcicek, E.E.; Louvi, A.; Bobadilla, N.A.; Gamba, G.; Lifton, R.P. WNK2 kinase is a novel regulator of essential neuronal cation-chloride cotransporters. Biol. Chem. 2011, 286, 30171–30180, doi:10.1074/jbc.M111.222893.
- Haas, M.; McBrayer, D.; Lytle, C. [Cl−] i-dependent phosphorylation of the Na-K-Cl cotransport protein of dog tracheal epithelial cells. Biol. Chem. 1995, 270, 28955–28961.
- Lytle, C.; Forbush 3rd, B. Regulatory phosphorylation of the secretory Na-K-Cl cotransporter: Modulation by cytoplasmic Cl. J. Physiol. Cell Physiol. 1996, 270, C437–C448.
- Cossins, A.; Weaver, Y.; Lykkeboe, G.; Nielsen, O. Role of protein phosphorylation in control of K flux pathways of trout red blood cells. J. Physiol. Cell Physiol. 1994, 267, C1641–C1650.
- Flatman, P.W.; Adragna, N.C.; Lauf, P.K. Role of protein kinases in regulating sheep erythrocyte K-Cl cotransport. J. Physiol. Cell Physiol. 1996, 271, C255–C263.
- Jennings, M.L.; Schulz, R.K. Okadaic acid inhibition of KCl cotransport. Evidence that protein dephosphorylation is necessary for activation of transport by either cell swelling or N-ethylmaleimide. Gen. Physiol. 1991, 97, 799–817.
- McCormick, J.A.; Ellison, D.H. The WNKs: Atypical protein kinases with pleiotropic actions. Rev. 2011, 91, 177–219.
- Arroyo, J.P.; Kahle, K.T.; Gamba, G. The SLC12 family of electroneutral cation-coupled chloride cotransporters. Asp. Med. 2013, 34, 288–298.
- Piala, A.T.; Moon, T.M.; Akella, R.; He, H.; Cobb, M.H.; Goldsmith, E.J. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Signal. 2014, 7, ra41, doi:10.1126/scisignal.2005050.
- Maruyama, J.; Kobayashi, Y.; Umeda, T.; Vandewalle, A.; Takeda, K.; Ichijo, H.; Naguro, I. Osmotic stress induces the phosphorylation of WNK4 Ser575 via the p38MAPK-MK pathway. Rep. 2016, 6, 18710, doi:10.1038/srep18710.
- Salihu, S.; Meor Azlan, N.; Josiah, S.; Wu, Z.; Wang, Y.; Zhang, J. Role of the cation-chloride-cotransporters in the circadian system. Asian J. Pharm. Sci. 2020, doi:1016/j.ajps.2020.10.003.
- Zhang, J.; Gao, G.; Begum, G.; Wang, J.; Khanna, A.R.; Shmukler, B.E.; Daubner, G.M.; de Los Heros, P.; Davies, P.; Varghese, J.; et al. Functional kinomics establishes a critical node of volume-sensitive cation-Cl(-) cotransporter regulation in the mammalian brain. Rep. 2016, 6, 35986, doi:10.1038/srep35986.
- Adragna, N.C.; Ravilla, N.B.; Lauf, P.K.; Begum, G.; Khanna, A.R.; Sun, D.; Kahle, K.T. Regulated phosphorylation of the K-Cl cotransporter KCC3 is a molecular switch of intracellular potassium content and cell volume homeostasis. Cell Neurosci. 2015, 9, 255, doi:10.3389/fncel.2015.00255.
- Rinehart, J.; Maksimova, Y.D.; Tanis, J.E.; Stone, K.L.; Hodson, C.A.; Zhang, J.; Risinger, M.; Pan, W.; Wu, D.; Colangelo, C.M. Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell 2009, 138, 525–536.
- Thastrup, J.O.; Rafiqi, F.H.; Vitari, A.C.; Pozo-Guisado, E.; Deak, M.; Mehellou, Y.; Alessi, D.R. SPAK/OSR1 regulate NKCC1 and WNK activity: Analysis of WNK isoform interactions and activation by T-loop trans-autophosphorylation. J. 2012, 441, 325–337, doi:10.1042/BJ20111879.
- Markkanen, M.; Ludwig, A.; Khirug, S.; Pryazhnikov, E.; Soni, S.; Khiroug, L.; Delpire, E.; Rivera, C.; Airaksinen, M.S.; Uvarov, P. Implications of the N-terminal heterogeneity for the neuronal K-Cl cotransporter KCC2 function. Brain Res. 2017, 1675, 87–101, doi:10.1016/j.brainres.2017.08.034.
- Vitari, A.C.; Thastrup, J.; Rafiqi, F.H.; Deak, M.; Morrice, N.A.; Karlsson, H.K.; Alessi, D.R. Functional interactions of the SPAK/OSR1 kinases with their upstream activator WNK1 and downstream substrate NKCC1. J. 2006, 397, 223–231, doi:10.1042/BJ20060220.
- Zhao, H.; Nepomuceno, R.; Gao, X.; Foley, L.M.; Wang, S.; Begum, G.; Zhu, W.; Pigott, V.M.; Falgoust, L.M.; Kahle, K.T.; et al. Deletion of the WNK3-SPAK kinase complex in mice improves radiographic and clinical outcomes in malignant cerebral edema after ischemic stroke. Cereb. Blood Flow Metab. 2017, 37, 550–563, doi:10.1177/0271678X16631561.
- Cuomo, O.; Vinciguerra, A.; Cerullo, P.; Anzilotti, S.; Brancaccio, P.; Bilo, L.; Scorziello, A.; Molinaro, P.; Di Renzo, G.; Pignataro, G. Ionic homeostasis in brain conditioning. Neurosci. 2015, 9, 277.
- Tao, D.; Liu, F.; Sun, X.; Qu, H.; Zhao, S.; Zhou, Z.; Xiao, T.; Zhao, C.; Zhao, M. Bumetanide: A review of its neuroplasticity and behavioral effects after stroke. Neurol. Neurosci. 2019, 37, 397–407.
- Jaenisch, N.; Witte, O.W.; Frahm, C. Downregulation of potassium chloride cotransporter KCC2 after transient focal cerebral ischemia. Stroke 2010, 41, e151–e159, doi:10.1161/STROKEAHA.109.570424.
- Wang, T.; Kumada, T.; Morishima, T.; Iwata, S.; Kaneko, T.; Yanagawa, Y.; Yoshida, S.; Fukuda, A. Accumulation of GABAergic neurons, causing a focal ambient GABA gradient, and downregulation of KCC2 are induced during microgyrus formation in a mouse model of polymicrogyria. Cortex 2014, 24, 1088–1101, doi:10.1093/cercor/bhs375.
- Yan, Y.; Dempsey, R.J.; Flemmer, A.; Forbush, B.; Sun, D. Inhibition of Na+–K+–Cl− cotransporter during focal cerebral ischemia decreases edema and neuronal damage. Brain Res. 2003, 961, 22–31.
- Yan, Y.; Dempsey, R.J.; Sun, D. Na+-K+-Cl− cotransporter in rat focal cerebral ischemia. Cereb. Blood Flow Metab. 2001, 21, 711–721.
- Chen, X.; Kintner, D.B.; Luo, J.; Baba, A.; Matsuda, T.; Sun, D. Endoplasmic reticulum Ca2+ dysregulation and endoplasmic reticulum stress following in vitro neuronal ischemia: Role of Na+-K+-Cl- J. Neurochem. 2008, 106, 1563–1576.
- Chen, H.; Sun, D. The role of Na–K–Cl co–transporter in cerebral ischemia. Res. 2005, 27, 280–286.
- Wu, D.; Zhang, G.; Zhao, C.; Yang, Y.; Miao, Z.; Xu, X. Interleukin-18 from neurons and microglia mediates depressive behaviors in mice with post-stroke depression. Brain Behav. Immun. 2020, 88, 411–420.
- Huang, L.-Q.; Zhu, G.-F.; Deng, Y.-Y.; Jiang, W.-Q.; Fang, M.; Chen, C.-B.; Cao, W.; Wen, M.-Y.; Han, Y.-L.; Zeng, H.-K. Hypertonic saline alleviates cerebral edema by inhibiting microglia-derived TNF-α and IL-1β-induced Na-K-Cl Cotransporter up-regulation. Neuroinflamm. 2014, 11, 1–20.
- Su, G.; Kintner, D.B.; Flagella, M.; Shull, G.E.; Sun, D. Astrocytes from Na+-K+-Cl− cotransporter-null mice exhibit absence of swelling and decrease in EAA release. J. Physiol. Cell Physiol. 2002, 282, C1147–C1160.
- Chen, H.; Luo, J.; Kintner, D.B.; Shull, G.E.; Sun, D. Na+-dependent chloride transporter (NKCC1)-null mice exhibit less gray and white matter damage after focal cerebral ischemia. Cereb. Blood Flow Metab. 2005, 25, 54–66.
- Lee, H.A.; Hong, S.H.; Kim, J.W.; Jang, I.S. Possible involvement of DNA methylation in NKCC1 gene expression during postnatal development and in response to ischemia. Neurochem. 2010, 114, 520–529.
- Bhuiyan, M.I.H.; Song, S.; Yuan, H.; Begum, G.; Kofler, J.; Kahle, K.T.; Yang, S.-S.; Lin, S.-H.; Alper, S.L.; Subramanya, A.R. WNK-Cab39-NKCC1 signaling increases the susceptibility to ischemic brain damage in hypertensive rats. Cereb. Blood Flow Metab. 2017, 37, 2780–2794.
- Hertz, L.; Xu, J.; Chen, Y.; Gibbs, M.E.; Du, T. Antagonists of the Vasopressin V1 Receptor and of the β1-Adrenoceptor Inhibit Cytotoxic Brain Edema in Stroke by Effects on Astrocytes-but the Mechanisms Differ. Neuropharmacol. 2014, 12, 308–323.
- Lu, K.-T.; Huang, T.-C.; Wang, J.-Y.; You, Y.-S.; Chou, J.-L.; Chan, M.W.; Wo, P.Y.; Amstislavskaya, T.G.; Tikhonova, M.A.; Yang, Y.-L. NKCC1 mediates traumatic brain injury-induced hippocampal neurogenesis through CREB phosphorylation and HIF-1α expression. Pflügers Arch. Eur. J. Physiol. 2015, 467, 1651–1661.
- Fu, P.; Tang, R.; Yu, Z.; Huang, S.; Xie, M.; Luo, X.; Wang, W. Bumetanide-induced NKCC1 inhibition attenuates oxygen–glucose deprivation-induced decrease in proliferative activity and cell cycle progression arrest in cultured OPCs via p-38 MAPKs. Brain Res. 2015, 1613, 110–119.
- Yu, Y.; Fu, P.; Yu, Z.; Xie, M.; Wang, W.; Luo, X. NKCC1 inhibition attenuates chronic cerebral hypoperfusion-induced white matter lesions by enhancing progenitor cells of oligodendrocyte proliferation. Mol. Neurosci. 2018, 64, 449–458.
- Mu, X.; Wang, H.; Cheng, X.; Yang, L.; Sun, X.; Qu, H.; Zhao, S.; Zhou, Z.; Liu, T.; Xiao, T. Inhibition of Nkcc1 promotes axonal growth and motor recovery in ischemic rats. Neuroscience 2017, 365, 83–93.
- Yuen, N.Y.; Chechneva, O.V.; Chen, Y.-J.; Tsai, Y.-C.; Little, L.K.; Dang, J.; Tancredi, D.J.; Conston, J.; Anderson, S.E.; O’Donnell, M.E. Exacerbated brain edema in a rat streptozotocin model of hyperglycemic ischemic stroke: Evidence for involvement of blood–brain barrier Na–K–Cl cotransport and Na/H exchange. Cereb. Blood Flow Metab. 2019, 39, 1678–1692.


