 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Božena Smolková | + 3073 word(s) | 3073 | 2021-01-19 15:42:14 |
Video Upload Options
Epigenetic dysregulation has been recognized as a critical factor contributing to the development of resistance against standard chemotherapy and to breast cancer progression via epithelial-to-mesenchymal transition. Although the efficacy of the first-generation epigenetic drugs (epi-drugs) in solid tumor management has been disappointing, there is an increasing body of evidence showing that epigenome modulation, in synergy with other therapeutic approaches, could play an important role in cancer treatment, reversing acquired therapy resistance.
1. Introduction
The most common cancer diagnosed among women is breast cancer (BC), the second leading cause of cancer deaths [1]. Besides well-studied genetic changes, epigenetic alterations, resulting in aberrant gene expression, are among the key contributors to breast carcinogenesis. Different mechanisms introduce and maintain epigenetic modifications, including DNA methylation, post-translational histone modifications, and non-coding RNA-mediated regulation [2]. Epithelial-mesenchymal transition (EMT) is a complex developmental program, which plays a crucial role in the hematogenous and lymphatic dissemination of tumors. EMT facilitates phenotypic metamorphosis of epithelial tumor cells into highly motile and more aggressive mesenchymal cells that can colonize distant organs. Moreover, this multistep process enables the generation of tumors with stem cell properties, which play a significant role in developing therapeutic resistance [3]. The reversibility of EMT, allowing circulating tumor cells (CTCs) to remain epithelial in their origin, endowing them with a potential to seed metastasis, supports the hypothesis about its epigenetic regulation [4]. EMT is triggered by extracellular signals, including extracellular matrix proteins and soluble growth factors, or by intracellular cues. It is mediated by a group of pleiotropic transcription factors (TFs), which control a heterogeneous network of epigenetic effectors, thus allowing potent gene expression changes [4]. This epigenetic plasticity not only permits dynamic regulation of expression but also offers numerous therapeutic opportunities.
Currently, BC treatment involves a multidisciplinary approach. Although the effectiveness of various therapeutic regimens has increased, resulting in reduced mortality [5], there are still many obstacles to overcome. These include serious side effects, hard-to-treat tumor subtypes, intratumoral heterogeneity, and at present incurable metastatic disease. The success achieved so far in treating hematological malignancies using epigenetic inhibitors has stimulated interest in their use to treat solid tumors. Promising preclinical results suggest that epigenetic drugs (epi-drugs) can sensitize resistant cancer cells to traditional approaches. Unfortunately, these results have not yet been confirmed by clinical studies, as the early-generation epi-drugs were basically broad-spectrum reprogrammers, causing large-scale gene expression changes. This "one size fits all" approach has mostly failed due to off-target effects, significant toxicities, the risk of large-scale epigenomic repatterning, and the lack of appropriate biomarkers for patient selection. However, this failure has led to the development of selective new-generation epi-drugs, which, together with precision medicine design, provide a new chance for epigenetic therapy of solid tumors [6][7].
Among the options that could contribute to successful clinical applications of epi-drugs are new technologies for safer and more efficient cancer cell epigenome modulation. Advances in nanotechnology and material science have provided a broad variety of more precise and safer nanoscale organic and inorganic nanomaterials for drug delivery (e.g., dendrimers, micelles, liposomes, gels, metal- and carbon-based nanomaterials). To date, several nanomaterials have been successfully studied and introduced in cancer treatment, and many others are undergoing clinical trials. Encapsulation by intelligent nanocarriers of antitumor drugs, conventional chemotherapeutics, epi-drugs, or both, can improve their solubility and stability by protecting the drugs from fast clearance and degradation, thus prolonging their half-life in the systemic circulation [8]. Nanocarriers can also be tuned to ensure targeted, controlled, and sustained release, thereby reducing toxicity [8][9][10]. Nanoscale size and unique physicochemical properties (e.g., shape, surface area, and charge) allow the accumulation of nanocarriers in the tumor mass due to the enhanced permeability and retention (EPR) effect, which is the basis of passive targeting [11]. The spatial and temporal heterogeneity of tumors is one of the limitations of therapeutic efficacy in passive targeting [12]. Functional surface modifications of nanocarriers by specific ligands (antibodies, aptamers, proteins, etc.) with a high affinity for particular receptors overexpressed on the tumor cells allows active targeting of drug delivery to the tumor mass, thereby increasing treatment efficacy and reducing side effects [13]. The biggest challenge to combat BC is to eliminate cancer stem cells (CSCs) that play a crucial role in metastasis and the development of multidrug resistance to therapy. Nanoscale delivery systems represent a promising tool for their eradication [14].
2. Molecular Pathology of Breast Cancer
The BC incidence rate varies from 27.9 per 100,000 people in Middle Africa to 92.6 per 100,000 in Western Europe [1]. The differences in incidence are attributed to different risk factors and the availability of improved imaging techniques for screening and diagnosis [15]. The five-year relative survival rate for women diagnosed with the regional disease was recently estimated at 86%, whereas it was 27% for those with metastatic disease [16]. Therapeutic resistance and metastatic potential are influenced by the heterogeneity of phenotypic and molecular characteristics [17][18][19][20]. While the luminal A subtype is considered a low-grade disease with a good prognosis and likely to benefit from endocrine therapy alone, luminal B tumors have a higher proliferation rate, worse prognosis, and patients require additional chemotherapy treatment. The human epidermal receptor 2 (HER2)-overexpressing tumors tend to grow faster and can have higher histological grade than luminal-like tumors, but generally, they are successfully treated by targeted anti-HER2 therapies. Triple-negative/basal-like BC is a histologically high-grade disease associated with a poor prognosis. Patients with this subtype do not benefit from targeted therapies, and the standard chemotherapy regimen is the only suitable therapeutic approach at present.
3. Breast Cancer Therapeutic Opportunities
3.1. The Biomarker-Directed Approach in BC Treatment
To overcome the limitations incurred by resistance mechanisms in the clinical management of advanced cancers, an increasing effort is being made towards biomarker-driven cancer treatments. This aims to identify important biomarkers capable of addressing tumor heterogeneity and effectively predicting a favorable clinical outcome as a response to a particular treatment. BC, in particular, is considered a family of distinct diseases with a varying molecular basis. Initial gene expression studies using cDNA microarrays have resulted in a classification of BC into five subtypes, establishing as major BC biomarkers estrogen receptor (ER), progesterone receptor (PR), and HER2 [21][22]. Therefore, currently, there is a mandatory need to define their expression status, lymph node involvement, and tumor size for all patients with invasive BC for therapy decision making. These markers are established in international guidelines as essential factors for the clinical management of primary BC patients [15]. However, the stratification of patients based solely on ER, PR, and HER2 expression has proven inefficient, unable to capture the substantial phenotypic complexity and heterogeneity of BC, thus stressing the need to integrate additional biomarkers for a more refined characterization. Ki-67, a non-histone nuclear protein, is used as a marker of cell proliferation. Ki-67 expression is significantly higher in malignant tissues with poorly differentiated tumor cells than in normal tissue and is thus used to assess tumor aggressiveness [23].
Traditional therapy of non-metastatic BC involves multidisciplinary strategies combining surgery, radiotherapy, neo-/adjuvant, endocrine, and targeted therapy [24]. For non-metastatic BC, the primary therapy approach consists of eradicating tumor and regional lymph nodes and preventing metastatic relapse. The first two goals are usually achieved by locoregional therapy that involves surgery and radiotherapy and/or neoadjuvant therapy in case of locally advanced disease. Prevention of metastatic relapse is achieved with systemic therapies that comprise anthracycline and taxane-based chemotherapy, anti-estrogen hormonal therapy, and anti-HER treatment, depending on receptor status [25].
In contrast to early BC, metastatic disease (stage IV), with common sites of spread in bones, brain, lung, and liver, is considered incurable, and the therapy aims to prolong life while minimizing symptoms or side effects. The combinations of endocrine, targeted therapy, chemotherapy, and immunotherapy can be administered to the metastatic patients, taking into account the tumor subtype, extent, and localization of the disease and the presence of specific molecular alterations. Beyond HER2 and ER/PR, new predictive biomarkers for targeted therapy in metastatic BC include BRCA1/2 and PI3KCA mutations for PARP and PI3KCA inhibitors, respectively, and PD-L1 expression and/or MSI status for immunotherapy. The effective new biological therapies like CDK4/6 or mTOR inhibitors are now emerging. However, we still lack predictive biomarkers for these treatments [26].
3.2. Precision Medicine Concept
The maturation of omic technologies as powerful molecular epidemiological screening tools has empowered the emergence of manifold predictive biomarker signatures. The integration of genomic and transcriptomic profiles of 2000 breast tumors from the METABRIC (Molecular Taxonomy of Breast Cancer International Consortium) cohort revealed ten BC subtypes, termed integrative clusters (IntClust/s) and characterized by distinct genomic drivers [27]. Currently, there are five main standardized genetic prognostic platforms for BC, aiming to assist decision on therapeutic options, mainly including hormone therapy, chemotherapy, and anti-HER2 treatment:
-
Oncotype DX provides prognostic information in terms of 10-year distant recurrence. It predicts the likelihood of adjuvant chemotherapy benefit in ER+ BC patients, based on the expression of a panel of 21 genes (16 cancer-related and five reference genes) [28].
-
Breast Cancer Index assesses the expression of 7 genes to predict the benefit from extended, adjuvant, endocrine therapy (Tamoxifen) in HR+ patients. It is a gene expression signature comprising two functional biomarker panels, the molecular grade index (MGI) and the two-gene ratio HOXB13/IL17BR (H/I), that evaluate tumor proliferation and estrogen signaling, respectively [29]. MGI is a gene expression assay, measuring the expression of five genes (BUB1B, CENPA, NEK2, RACGAP1, RRM2) related to histological grade and tumor progression, which recapitulates tumor grade and can predict the clinical outcome with high performance [30].
-
EndoPredict (Myriad Genetics, Inc., Salt Lake City, UT, USA) is a genomic test for people newly diagnosed with early-stage, ER+, HER2-negative BC (node-negative). It assesses the expression of 12 genes (8 target genes, 3 normalization genes, and 1 control gene) to predict response to chemotherapy [31].
-
MammaPrint (Agendia, Irvine, CA, USA) is a 70-gene signature test that predicts the clinical outcome/response to chemotherapy in ER+ early-stage BC [32].
-
Prosigna Breast Cancer Prognostic Gene Signature Assay (Nanostring, Seattle, WA, USA), formerly PAM50, assesses Tamoxifen response for HR+ BC patients based on the expression of 58 genes after 5 years of hormonal therapy treatment in postmenopausal women [33].
Besides Prosigna, Nanostring has developed a more extensive assay, the human nCounter Breast Cancer 360 panel, which comprises 776 genes across 23 key BC pathways and processes. Results are grouped in 48 signatures across 13 categories, measuring biological variables crucial to BC tumor biology. This panel has been developed for the evaluation of diverse BC aspects, including BC subtyping (luminal A/B, HER2-enriched, basal-like, triple-negative), expression of BC receptors and signaling (ESR1, PGR, ERBB2, AR, PTEN, CDK4, CDK6), mutational content (HRD, BRCA, P53), markers for tumor proliferation, apoptosis and differentiation (FOXA1, SOX2), cell adhesion (claudin), and immunity (chemokines, TGF-β, PD-1). Although a detailed description of the nCounterBC 360 panel exceeds this review’s scope, it contains a vast number of gene expression markers and indicates the intense molecular heterogeneity characterizing the diversity of BC phenotypes. Therefore, investigating the epigenetic landscape of BC may provide an additional layer of information that could improve our fundamental understanding of BC's molecular complexity and the putative rational development of more effective and precise treatments.
Finally, various studies have identified and suggested epigenetic modifications and regulators as prognostic biomarkers for BC [34][35][36]. In this context, miRNAs have been considered a pool of highly potent biomarkers, as they have been linked to the identification of distinct molecular subtypes and tumor-related processes. miRNA expression profiling was successfully employed to classify the breast tumors as luminal A, luminal B, basal-like, HER2+, and normal-like BC [37]. Aberrant expression of miRNAs has also been correlated with clinical features, such as angiogenesis, metastasis, and EMT [38]. Besides, several lncRNAs have been reported as promising biomarkers for prognosis, diagnosis, and therapy [39]. Overall, there is cumulative evidence focusing on the systematic screening of epigenetic signals as a promising area for the discovery of novel molecular BC biomarkers, combining sensitivity, specificity, and robustness, with a potentially decisive impact on improving the quality of BC treatments in the context of precision medicine.
3.3. Potential of Epigenetic Therapy
The role of epigenetics in cancer initiation and progression, including its contribution to the development of innate and acquired resistance to several therapeutic regimens, has led to the scientific effort to reverse the aberrant cancer epigenome [40]. The lack of knowledge about subtype-specific epigenome signaling pathways, and missing patient-specific epigenetic biomarker profiles, are currently the main challenges hampering the wider clinical application of epigenetic agents in the treatment of solid cancers [41]. Over the last decade, several epi-drugs have received US Food and Drug Administration (FDA) approval for the treatment of blood-borne cancers: 5-azacytidine (AZA, Vidaza®), 5-aza-2'-deoxycytidine (decitabine, DAC, Dacogen®), vorinostat (VOR, SAHA, Zolinza®), romidepsin (FK228, Istodax®), belinostat (Beleodaq®), panobinostat (Farydak®), and chidamide (Epidaza®) (Figure 1) [42][43][44]. However, except for tazemetostat (Tazverik®), approved by FDA in January 2020 for metastatic or locally advanced epithelioid sarcoma, there is no epigenetic therapy approved for solid tumors, which are considered more epigenetically complex. Moreover, they exhibit abnormal vascularization, a specific tumor microenvironment, and more differentiated cells with decreased epigenetic reprogramming [7][45].
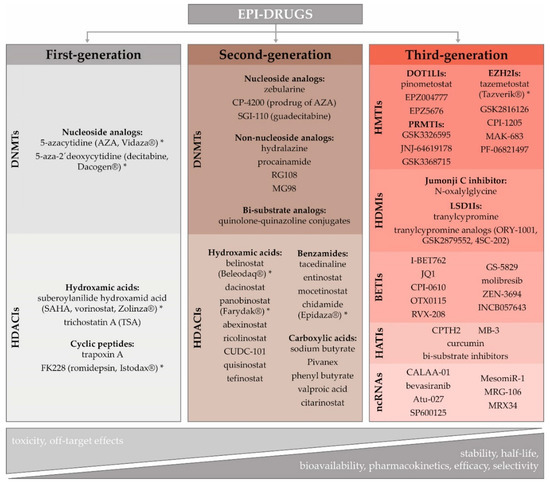
Figure 1. Different categories of epi-drugs, assessed in preclinical studies and clinical trials. Eight of them (indicated by asterisks) were approved to treat several human malignancies (modified from [7][46][47]). Abbreviations: DNMTIs-DNA methyltransferase inhibitors; HDACIs-histone deacetylase inhibitors; HMTIs-histone methyltransferase inhibitors; HDMIs-histone demethylase inhibitors; BETIs-bromodomain and extra-terminal domain inhibitors; HATIs-histone acetyltransferase inhibitors; ncRNAs-non-coding RNAs; DOT1LIs-DOT1-like histone lysine methyltransferase inhibitors; EZH2Is-enhancer of zeste homolog 2 inhibitors; PRMTIs-protein arginine methyltransferase inhibitor; LSD1Is-lysine-specific histone demethylase 1A inhibitors.
Rational epi-drug discovery using validated targets is a recent phenomenon. During early efforts, epi-drug development has been based on the demonstration of efficacy and phenotypic observations rather than on knowledge of their molecular targets. The timeline and key events influencing epi-drug development, including challenges and opportunities associated with their implementation in clinics, have recently been reviewed in-depth by Ganesan and colleagues [46]. As a detailed characterization of epi-drugs is beyond this paper’s scope, we provide only their basic characteristics and classification (Figure 1).
Given that overexpression of DNMTs and HDACs are considered the critical factors in carcinogenesis, demethylating agents and HDAC inhibitors (HDACIs) seem to be promising anticancer drugs [47][48][49]. Cytidine analogs, AZA and DAC, were the first DNMT inhibitors (DNMTIs) approved by the FDA in 2004 for the treatment of myelodysplastic syndrome and acute myeloid leukemia [50]. Although they were initially defined as cytotoxic agents, their therapeutic properties were achieved at lower doses and with prolonged exposure [51]. In general, HDACIs block histone deacetylation, causing reactivation of tumor suppressor genes that can inhibit cancer cell proliferation. Moreover, they have been shown to induce cancer cell death at concentrations to which normal cells are relatively resistant [52][53]. Chemically, HDACIs are classified into different subgroups: carboxylic acids, benzamides, cyclic peptides, and hydroxamic acids [54]. Similar to DNMTs, the first-generation HDACIs were characterized by poor bioavailability, low stability, and short half-life.
The development of second-generation epi-drugs has, therefore, been aimed to circumvent these shortcomings. Guadecitabine, a second-generation DNMTI, has a novel molecular structure, which prolongs its in vivo half-life, and increases efficacy [55]. As nucleoside analogs require active DNA synthesis to incorporate them into the DNA, their use is limited in hypoproliferative cancers and could be a major obstacle in the therapy of solid tumors [56]. Their common side effects could be avoided using non-nucleoside analogs, such as hydralazine, procainamide, RG108, and MG98 [57]. Recently, more efficient bi-substrate analogs have become potent DNMTIs [46]. Although the second-generation HDACIs, such as hydroxamic acid, belinostat, panobinostat, chidamide, or valproic acid, possess improved pharmacological properties, they achieved limited efficacy as single agents. However, their combination with other therapeutic approaches has allowed new avenues of their clinical investigation [7].
The principle of precision medicine is now being applied to the development and the use of third-generation epi-drugs, defined by a high degree of selectivity. This family includes HMT inhibitors (e.g., EZH2, DOT1-like histone-lysine methyltransferase (DOT1L), G9a and PRMT inhibitors), HDM inhibitors (e.g., LSD1 or Jumonji C domain inhibitors), BET inhibitors (BETIs) and HAT inhibitors (HATIs) [7][58]. HMTIs are emerging therapies targeting specific modifications. For example, it has been found that mutations in lymphomas activate the H3K27 histone methyltransferase EZH2, leading to disease progression. Therefore, the EZH2I can selectively target and induce cell death in cell lines with these mutations [59]. DOT1L is the only histone methyltransferase that targets the histone H3 lysine 79 (H3K79) residue [60]. Aberrant H3K79 methylation is associated with aggressive mixed-lineage leukemia and poor patient prognosis in lung, colorectal, and BCs [61]. This suggests that pharmacological inhibition of DOT1L can have therapeutic potential in several cancer types. The H3K4 and H3K9 demethylase enzyme LSD1 has an essential regulatory role in cell proliferation [62]. Its overexpression in several tumors has been correlated with a worse prognosis [63]. LSD1 inhibition may slow down cell growth in LSD1-overexpressing tumor cells. BETIs, JQ1, and I-BET762, are cell-permeable agents that reversibly and specifically bind the bromodomain proteins, thus impeding their interaction with acetylated histone lysine residues. It has been shown that they inhibit proliferation and induce apoptosis in various cancer cells [64]. HATIs include peptides, small molecules derived from natural products (e.g., curcumin), and synthetic molecules [65]. Peptide-CoA bisubstrate inhibitors mimic the formation of the substrate and cofactor complex binding to the HAT enzyme [58]. Well-conceived computational strategies and new screening platforms will be needed to predict loci specific epi-drugs sensitivities.
The ncRNAs, like miRNAs or siRNAs, with their power to selectively "switch-off" specific cancer genes, are attractive targets for the development of personalized cancer therapy. The main hindrance to the implementation of ncRNA-based therapy in clinical practice is the absence of effective delivery systems that can protect the RNA molecules from fast nuclease degradation before delivering them into the target cells' cytoplasm [66]. There are two different possibilities to use miRNAs as therapeutic agents; substitution of depleted miRNAs (MRX34, miR34a replacement) and inhibition of overexpressed miRNAs by antagonistic oligonucleotides [67]. The inhibitor of miR-155, MRG-106, has been successfully investigated in phase I clinical trials for the treatment of hematological malignancies [68]. The miRNA-based therapeutic strategy also has great potential to regulate lncRNAs. The siRNA-mediated silencing by oligonucleotide inhibitors results in the inhibition of lncRNA-protein interactions and secondary structure changes, thus competing for their binding partners [69]. Down-regulation of cancer-related genes by siRNAs, e.g., CALAA-01 (targeting RRM2), Atu-027 (targeting PKN3), has been assessed in phase I/II clinical trials [66].
Although several ongoing clinical trials, including epi-drugs, exist on a wide range of diseases, many obstacles remain to be resolved. Among them are enzyme isoform selectivity, dual substrates, multimeric enzyme complexes involved in epigenetic regulations, high-order chromatin structure, functional effects of inhibition, and off-target effects. Other challenges are the pharmacology of the compounds, doses to be used, therapeutic regimens or duration of the treatment, and patient selection.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424, doi:10.3322/caac.21492.
- Dworkin, A.M.; Huang, T.H.M.; Toland, A.E. Epigenetic alterations in the breast: Implications for breast cancer detection, prognosis and treatment. Semin. Cancer Biol. 2009, 19, 165–171, doi:10.1016/j.semcancer.2009.02.007.
- Roche, J. The Epithelial-to-Mesenchymal Transition in Cancer. Cancers 2018, 10, 52, doi:10.3390/cancers10020052.
- Sun, L.; Fang, J. Epigenetic regulation of epithelial-mesenchymal transition. Cell. Mol. Life Sci. 2016, 73, 4493–4515, doi:10.1007/s00018-016-2303-1.
- Moo, T.A.; Sanford, R.; Dang, C.; Morrow, M. Overview of Breast Cancer Therapy. PET Clin. 2018, 13, 339–354, doi:10.1016/j.cpet.2018.02.006.
- Lu, Y.; Chan, Y.T.; Tan, H.Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic regulation in human cancer: The potential role of epi-drug in cancer therapy. Mol. Cancer 2020, 19, 79, doi:10.1186/s12943-020-01197-3.
- Morel, D.; Jeffery, D.; Aspeslagh, S.; Almouzni, G.; Postel-Vinay, S. Combining epigenetic drugs with other therapies for solid tumours—Past lessons and future promise. Nat. Rev. Clin. Oncol. 2020, 17, 91–107, doi:10.1038/s41571-019-0267-4.
- Magro, M.; Venerando, A.; Macone, A.; Canettieri, G.; Agostinelli, E.; Vianello, F. Nanotechnology-Based Strategies to Develop New Anticancer Therapies. Biomolecules 2020, 10, 735, doi:10.3390/biom10050735.
- Jain, V.; Kumar, H.; Anod, H.V.; Chand, P.; Gupta, N.V.; Dey, S.; Kesharwani, S.S. A review of nanotechnology-based approaches for breast cancer and triple-negative breast cancer. J. Control. Release 2020, 326, 628–647, doi:10.1016/j.jconrel.2020.07.003.
- Thakur, V.; Kutty, R.V. Recent advances in nanotheranostics for triple negative breast cancer treatment. J. Exp. Clin. Cancer Res. 2019, 38, 430, doi:10.1186/s13046-019-1443-1.
- Hobbs, S.K.; Monsky, W.L.; Yuan, F.; Roberts, W.G.; Griffith, L.; Torchilin, V.P.; Jain, R.K. Regulation of transport pathways in tumor vessels: Role of tumor type and microenvironment. Proc. Natl. Acad. Sci. USA 1998, 95, 4607–4612, doi:10.1073/pnas.95.8.4607.
- Ernsting, M.J.; Murakami, M.; Roy, A.; Li, S.D. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J. Control. Release 2013, 172, 782–794, doi:10.1016/j.jconrel.2013.09.013.
- Navya, P.N.; Kaphle, A.; Srinivas, S.P.; Bhargava, S.K.; Rotello, V.M.; Daima, H.K. Current trends and challenges in cancer management and therapy using designer nanomaterials. Nano Converg. 2019, 6, 23, doi:10.1186/s40580-019-0193-2.
- Gao, Y.; Tang, M.; Leung, E.; Svirskis, D.; Shelling, A.; Wu, Z. Dual or multiple drug loaded nanoparticles to target breast cancer stem cells. RSC Adv. 2020, 10, 19089–19105.
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66, doi:10.1038/s41572-019-0111-2.
- Howlader, N.; Noone, A.; Krapcho, M. SEER Cancer Statistics Review, 1975–2014, Based on November 2016 SEER Data Submission; National Cancer Institute: Bethesda, MD, USA, 2017.
- Goncalves, H., Jr.; Guerra, M.R.; Duarte Cintra, J.R.; Fayer, V.A.; Brum, I.V.; Bustamante Teixeira, M.T. Survival Study of Triple-Negative and Non-Triple-Negative Breast Cancer in a Brazilian Cohort. Clin. Med. Insights Oncol. 2018, 12, 1179554918790563, doi:10.1177/1179554918790563.
- Hinohara, K.; Polyak, K. Intratumoral heterogeneity: More than just mutations. Trends Cell Biol. 2019, 29, 569–579, doi:10.1016/j.tcb.2019.03.003.
- Koren, S.; Bentires-Alj, M. Breast Tumor Heterogeneity: Source of Fitness, Hurdle for Therapy. Mol. Cell 2015, 60, 537–546, doi:10.1016/j.molcel.2015.10.031.
- Turashvili, G.; Brogi, E. Tumor Heterogeneity in Breast Cancer. Front. Med. 2017, 4, 227, doi:10.3389/fmed.2017.00227.
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752, doi:10.1038/35021093.
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874.
- Li, L.T.; Jiang, G.; Chen, Q.; Zheng, J.N. Ki67 is a promising molecular target in the diagnosis of cancer. Mol. Med. Rep. 2015, 11, 1566–1572.
- Chew, H.K. Adjuvant therapy for breast cancer: Who should get what? West. J. Med. 2001, 174, 284–287, doi:10.1136/ewjm.174.4.284.
- Cardoso, F.; Kyriakides, S.; Ohno, S.; Penault-Llorca, F.; Poortmans, P.; Rubio, I.T.; Zackrisson, S.; Senkus, E. Early breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 1674, doi:10.1093/annonc/mdz189.
- Cardoso, F.; Senkus, E.; Costa, A.; Papadopoulos, E.; Aapro, M.; André, F.; Harbeck, N.; Aguilar Lopez, B.; Barrios, C.H.; Bergh, J.; et al. 4th ESO-ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC 4)†. Ann. Oncol. 2018, 29, 1634–1657, doi:10.1093/annonc/mdy192.
- Curtis, C.; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352.
- Paik, S.; Shak, S.; Tang, G.; Kim, C.; Baker, J.; Cronin, M.; Baehner, F.L.; Walker, M.G.; Watson, D.; Park, T. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med. 2004, 351, 2817–2826.
- Bartlett, J.; Sgroi, D.; Treuner, K.; Zhang, Y.; Ahmed, I.; Piper, T.; Salunga, R.; Brachtel, E.; Pirrie, S.; Schnabel, C. Breast Cancer Index and prediction of benefit from extended endocrine therapy in breast cancer patients treated in the Adjuvant Tamoxifen—To Offer More?(aTTom) trial. Ann. Oncol. 2019, 30, 1776–1783.
- Ma, X.-J.; Salunga, R.; Dahiya, S.; Wang, W.; Carney, E.; Durbecq, V.; Harris, A.; Goss, P.; Sotiriou, C.; Erlander, M. A five-gene molecular grade index and HOXB13: IL17BR are complementary prognostic factors in early stage breast cancer. Clin. Cancer Res. 2008, 14, 2601–2608.
- Simon, R.M.; Paik, S.; Hayes, D.F. Use of archived specimens in evaluation of prognostic and predictive biomarkers. J. Natl. Cancer Inst. 2009, 101, 1446–1452.
- Cardoso, F.; van’t Veer, L.J.; Bogaerts, J.; Slaets, L.; Viale, G.; Delaloge, S.; Pierga, J.-Y.; Brain, E.; Causeret, S.; DeLorenzi, M. 70-gene signature as an aid to treatment decisions in early-stage breast cancer. N. Engl. J. Med. 2016, 375, 717–729.
- Jensen, M.-B.; Lænkholm, A.-V.; Nielsen, T.O.; Eriksen, J.O.; Wehn, P.; Hood, T.; Ram, N.; Buckingham, W.; Ferree, S.; Ejlertsen, B. The Prosigna gene expression assay and responsiveness to adjuvant cyclophosphamide-based chemotherapy in premenopausal high-risk patients with breast cancer. Breast Cancer Res. 2018, 20, 79.
- Bae, Y.K.; Brown, A.; Garrett, E.; Bornman, D.; Fackler, M.J.; Sukumar, S.; Herman, J.G.; Gabrielson, E. Hypermethylation in histologically distinct classes of breast cancer. Clin. Cancer Res. 2004, 10, 5998–6005.
- De Oca, R.M.; Gurard-Levin, Z.A.; Berger, F.; Rehman, H.; Martel, E.; Corpet, A.; de Koning, L.; Vassias, I.; Wilson, L.O.; Meseure, D. The histone chaperone HJURP is a new independent prognostic marker for luminal A breast carcinoma. Mol. Oncol. 2015, 9, 657–674.
- Roessler, J.; Ammerpohl, O.; Gutwein, J.; Steinemann, D.; Schlegelberger, B.; Weyer, V.; Sariyar, M.; Geffers, R.; Arnold, N.; Schmutzler, R. The CpG island methylator phenotype in breast cancer is associated with the lobular subtype. Epigenomics 2015, 7, 187–199.
- Blenkiron, C.; Goldstein, L.D.; Thorne, N.P.; Spiteri, I.; Chin, S.-F.; Dunning, M.J.; Barbosa-Morais, N.L.; Teschendorff, A.E.; Green, A.R.; Ellis, I.O. MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Biol. 2007, 8, R214.
- Braicu, C.; Raduly, L.; Morar-Bolba, G.; Cojocneanu, R.; Jurj, A.; Pop, L.-A.; Pileczki, V.; Ciocan, C.; Moldovan, A.; Irimie, A. Aberrant miRNAs expressed in HER-2 negative breast cancers patient. J. Exp. Clin. Cancer Res. 2018, 37, 257.
- Qi, P.; Du, X. The long non-coding RNAs, a new cancer diagnostic and therapeutic gold mine. Mod. Pathol. 2013, 26, 155–165.
- Guo, M.; Peng, Y.; Gao, A.; Du, C.; Herman, J.G. Epigenetic heterogeneity in cancer. Biomark Res. 2019, 7, 23, doi:10.1186/s40364-019-0174-y.
- Valdespino, V.; Valdespino, P.M. Potential of epigenetic therapies in the management of solid tumors. Cancer Manag. Res. 2015, 7, 241–251, doi:10.2147/cmar.S70358.
- Jones, P.A.; Issa, J.P.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641, doi:10.1038/nrg.2016.93.
- Ito, Y.; Makita, S.; Tobinai, K. Development of new agents for peripheral T-cell lymphoma. Expert Opin. Biol. Ther. 2019, 19, 197–209, doi:10.1080/14712598.2019.1572746.
- Roberti, A.; Valdes, A.F.; Torrecillas, R.; Fraga, M.F.; Fernandez, A.F. Epigenetics in cancer therapy and nanomedicine. Clin. Epigenetics 2019, 11, 81, doi:10.1186/s13148-019-0675-4.
- Ramachandran, S.; Ient, J.; Göttgens, E.L.; Krieg, A.J.; Hammond, E.M. Epigenetic Therapy for Solid Tumors: Highlighting the Impact of Tumor Hypoxia. Genes 2015, 6, 935–956, doi:10.3390/genes6040935.
- Ganesan, A.; Arimondo, P.B.; Rots, M.G.; Jeronimo, C.; Berdasco, M. The timeline of epigenetic drug discovery: From reality to dreams. Clin. Epigenetics 2019, 11, 174, doi:10.1186/s13148-019-0776-0.
- Gnyszka, A.; Jastrzebski, Z.; Flis, S. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer Res. 2013, 33, 2989–2996.
- Bohl, S.R.; Bullinger, L.; Rücker, F.G. Epigenetic therapy: Azacytidine and decitabine in acute myeloid leukemia. Expert Rev. Hematol. 2018, 11, 361–371, doi:10.1080/17474086.2018.1453802.
- Roche, J.; Bertrand, P. Inside HDACs with more selective HDAC inhibitors. Eur. J. Med. Chem. 2016, 121, 451–483, doi:10.1016/j.ejmech.2016.05.047.
- Ghasemi, S. Cancer’s epigenetic drugs: Where are they in the cancer medicines? Pharm. J. 2020, 20, 367–379.
- Filippova, E.V.; Zemaitaitis, B.; Aung, T.; Wolfe, A.J.; Anderson, W.F. Structural Basis for DNA Recognition by the Two-Component Response Regulator RcsB. mBio 2018, 9, e01993-17, doi:10.1128/mBio.01993-17.
- Marks, P.A.; Dokmanovic, M. Histone deacetylase inhibitors: Discovery and development as anticancer agents. Expert Opin. Investig. Drugs 2005, 14, 1497–1511, doi:10.1517/13543784.14.12.1497.
- Montalvo-Casimiro, M.; González-Barrios, R.; Meraz-Rodriguez, M.A.; Juárez-González, V.T.; Arriaga-Canon, C.; Herrera, L.A. Epidrug Repurposing: Discovering New Faces of Old Acquaintances in Cancer Therapy. Front. Oncol. 2020, 10, 2461, doi:10.3389/fonc.2020.605386.
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39, doi:10.1172/jci69738.
- Garcia-Manero, G.; Roboz, G.; Walsh, K.; Kantarjian, H.; Ritchie, E.; Kropf, P.; O’Connell, C.; Tibes, R.; Lunin, S.; Rosenblat, T.; et al. Guadecitabine (SGI-110) in patients with intermediate or high-risk myelodysplastic syndromes: Phase 2 results from a multicentre, open-label, randomised, phase 1/2 trial. Lancet Haematol. 2019, 6, e317–e327, doi:10.1016/s2352-3026(19)30029-8.
- Graça, I.; Pereira-Silva, E.; Henrique, R.; Packham, G.; Crabb, S.J.; Jerónimo, C. Epigenetic modulators as therapeutic targets in prostate cancer. Clin. Epigenetics 2016, 8, 98, doi:10.1186/s13148-016-0264-8.
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62, doi:10.1038/s41392-019-0095-0.
- Wapenaar, H.; Dekker, F.J. Histone acetyltransferases: Challenges in targeting bi-substrate enzymes. Clin. Epigenetics 2016, 8, 59, doi:10.1186/s13148-016-0225-2.
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Della Pietra, A., III; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112, doi:10.1038/nature11606.
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-Lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018, 131, 2661–2669, doi:10.1182/blood-2017-12-818948.
- Wong, M.; Polly, P.; Liu, T. The histone methyltransferase DOT1L: Regulatory functions and a cancer therapy target. Am. J. Cancer Res. 2015, 5, 2823–2837.
- Wojtala, M.; Dąbek, A.; Rybaczek, D.; Śliwińska, A.; Świderska, E.; Słapek, K.; El-Osta, A.; Balcerczyk, A. Silencing Lysine-Specific Histone Demethylase 1 (LSD1) Causes Increased HP1-Positive Chromatin, Stimulation of DNA Repair Processes, and Dysregulation of Proliferation by Chk1 Phosphorylation in Human Endothelial Cells. Cells 2019, 8, 1212, doi:10.3390/cells8101212.
- Majello, B.; Gorini, F.; Saccà, C.D.; Amente, S. Expanding the Role of the Histone Lysine-Specific Demethylase LSD1 in Cancer. Cancers 2019, 11, 324, doi:10.3390/cancers11030324.
- Alqahtani, A.; Choucair, K.; Ashraf, M.; Hammouda, D.M.; Alloghbi, A.; Khan, T.; Senzer, N.; Nemunaitis, J. Bromodomain and extra-terminal motif inhibitors: A review of preclinical and clinical advances in cancer therapy. Future Sci. OA 2019, 5, Fso372, doi:10.4155/fsoa-2018-0115.
- Luan, Y.; Ngo, L.; Han, Z.; Wang, X.; Qu, M.; Zheng, Y.G. Histone acetyltransferases: Enzymes, assays, and inhibitors. In Epigenetic Technological Applications; Elsevier: Amsterdam, The Netherlands, 2015; pp. 291–317.
- Meseure, D.; Drak Alsibai, K.; Nicolas, A.; Bieche, I.; Morillon, A. Long Noncoding RNAs as New Architects in Cancer Epigenetics, Prognostic Biomarkers, and Potential Therapeutic Targets. BioMed Res. Int. 2015, 2015, 320214, doi:10.1155/2015/320214.
- Baumann, V.; Winkler, J. miRNA-based therapies: Strategies and delivery platforms for oligonucleotide and non-oligonucleotide agents. Future Med. Chem. 2014, 6, 1967–1984, doi:10.4155/fmc.14.116.
- Takahashi, R.-U.; Prieto-Vila, M.; Kohama, I.; Ochiya, T. Development of miRNA-based therapeutic approaches for cancer patients. Cancer Sci. 2019, 110, 1140–1147, doi:10.1111/cas.13965.
- Ma, L.; Chua, M.S.; Andrisani, O.; So, S. Epigenetics in hepatocellular carcinoma: An update and future therapy perspectives. World J. Gastroenterol. 2014, 20, 333–345, doi:10.3748/wjg.v20.i2.333.


