 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mirko Pesce | + 2531 word(s) | 2531 | 2021-01-19 10:39:18 | | | |
| 2 | Vivi Li | Meta information modification | 2531 | 2021-01-20 08:54:17 | | |
Video Upload Options
Neonatal hypoxic-ischemic (HI) brain injury is one of the major drawbacks of mortality and causes significant short/long-term neurological dysfunction in newborn infants worldwide. To date, due to multifunctional complex mechanisms of brain injury, there is no well-established effective strategy to completely provide neuroprotection. Although therapeutic hypothermia is the proven treatment for hypoxic-ischemic encephalopathy (HIE), it does not completely chang outcomes in severe forms of HIE. Therefore, there is a critical need for reviewing the effective therapeutic strategies to explore the protective agents and methods. In recent years, it is widely believed that there are neuroprotective possibilities of natural compounds extracted from plants against HIE. These natural agents with the anti-inflammatory, anti-oxidative, anti-apoptotic, and neurofunctional regulatory properties exhibit preventive or therapeutic effects against experimental neonatal HI brain damage.
1. Introduction
Hypoxia-ischemia (HI) is regarded as one of the major drawbacks of brain damage in the neonates and infants. Neonatal hypoxic-ischemic brain damage (HIBD) (synonymous with hypoxic-ischemic encephalopathy (HIE)) is the main drawback of newborn deaths and irretrievable and long-lasting neurodevelopmental disabilities in the sufferers [1]. HIE complications estimate 23% of infant deaths all over the world, and have impacted 0.7–1.2 million newborns per annum [2]. Cerebral palsy, epilepsy, mental retardation, motor and cognitive deficits, learning and behavioral disabilities, and other severe neurological disorders were regularly observed in the sufferers based on brain injury grade [3][4]. In infants with mild HIE, the death probability is 10% and the risk of neurodevelopmental disorders is 30%, while 60% of infants with severe HIE are at risk of death and all sufferers risk exposure to remarkable disabilities.
The neonatal HI (NHI) incidence is near 1–8 of every one thousand live full-term births in developed countries, and nearly 25 in one thousand live full-term births in developing countries [5][6]. Lately, advanced neonatology has evidenced a great improvement in survival of seriously premature newborns. The rate of incidence in the very low birth weight infants is 60% of all live births.
NHI commonly occurs as a result of prenatal or birth asphyxia near the time of birth [7][8]. The ordinary birth process that is correlated with asphyxia and severe asphyxia may affect a pre-existing normal brain, prompting acute encephalopathy. Severe asphyxia can arise for numerous causes, including abnormal uterine contractions, abruption of the placenta, umbilical cord compression, or failure of the neonate to initiate breathing successfully. Infection, growth retardation, and fever can enhance the brain sensitivity to nonpathogenic asphyxia. On the other hand, the prematurity subject is even more complicated which is frequently concomitant with impaired fetal growth and infection. Also, the general handling of the severe premature infants causes bases of postnatal insults. The most common form of perinatal injury is systemic asphyxia, which impacts the neonate/fetus cardiovascular system and results in a hypoperfusion by hypoxemic blood. Consequently, such injury is discussed to be reperfusion/hypoperfusion damage [9].
On the cellular status, HI actuates the activation of complex biochemical reactions, consisting of switching from oxidative to anaerobic metabolism, cellular energy failure, excitotoxicity, dysfunction of mitochondria, overload of intracellular calcium, production of free radicals, and inflammatory responses, which results in significant neuronal impairment and ultimately contributes to change in degrees of brain infarction, BBB (blood–brain barrier) dysfunction, and brain edema, finally resulting in irreversible brain injury.
The neurologic disorders extremely dwindle the quality of life in HIBD kids and escalate the socioeconomic burden on caregivers, families, and society. Currently, hypothermia is the only standard treatment for neonatal HIBD for full-term infants with moderate to severe HIE [10]. By this treatment, infants with the most severe type of HIE might not be saved, and this strategy involves the risk of severe disability or death, as nearly 50% of treated cases still die [11], and as many as 1/3 to 1/2 of sufferers demonstrate low Intelligence Quotient (IQ) at 6 to 7 years of age or constant neurologic abnormalities [12][13]. Therefore, more effective neuroprotective strategies are required. In this respect, several agents have been investigated to save the brain from irreversible damage or to postpone the pathological process, such as Erythropoietin, Melatonin, Metformin, stem cell therapy, etc., but only a few agents have been used in clinical studies [14]. Therefore, a search to explore better neuroprotective agents is under way.
2. Pathophysiology of Neonatal Hypoxic-Ischemic Brain Damage
2.1. Excitatory
In fact, HIE is caused by a restriction in blood supply (ischemia) and deprivation of adequate oxygen supply (hypoxia), which causes a switch to anaerobic metabolism. Anaerobic metabolism leads to depletion of adenosine triphosphate (ATP) and accumulation of lactic acid. This condition is known as primary energy failure, and occurs immediately after initial HI insult. At the cellular level, energy failure and ATP reduction results in failure of the cell membrane ion in pumping and consequently in flow of sodium and calcium ions accompanied by water flow into the cell. Following the process, the cytotoxic edema is eventuated due to the water flow which might lead to cellular swelling and cell lysis [9]. The following membrane depolarization unrolls voltage-sensitive calcium channels and leads to excessive calcium influx. Increase in intracellular Ca2+ leads to production and release of excitatory amino acids, particularly glutamate. In the following stage, excess glutamate activates receptors such as α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-D-aspartate (NMDA) receptors [15]. Activation of these receptors additionally promotes influx of Ca2+ into the affected cells. Finally, the rise in intracellular Ca2+ promotes production of reactive oxygen species (ROS), release of pro-radicals (such as free ions), and synthesis of excessive nitric oxide (NO) by neuronal NO synthase (nNOS), which is modulated by the NMDA receptors [15]. Furthermore, activation of lipases, proteases, and endonucleases are increased, which causes a release of fatty acids and membrane damage, and triggers a cascade reaction series which leads to cell death [15].
2.2. Free Radical Toxicity
Reoxygenation and reperfusion oxygen availability lead to the initial ischemic tissue and more ROS generation by mitochondria (via the electron transport chain) and damage integrity of membrane and organelle’s function. These activities lead to mitochondrial dysfunction which in turn can trigger ROS-induced ROS generation. Furthermore, hypoxanthine and pro-radicals such as iron cause large amounts of ROS formation. Mitochondrial impairment is one of the main factors involved in the apoptotic pathway of cell death. The second phase of injury is known as reperfusion injury and occurs hours later [15].
2.3. Inflammation
In addition to the described mechanisms, damaged neurons and activated endothelium produce various cytokines, including interleukins (IL-1, IL-6, IL-8, IL-10) and tumor necrosis factor (TNF)-α, which activate inflammatory response and trigger biochemical pathways that lead to secondary energy failure and a delay in neuronal cell death [15]. Secondary energy failure generally eventuates 6 to 48 h after initial HI insult and delays necrotic and apoptotic processes, that might continue up to days or weeks after birth [15]. Also, a delay in brain damage is associated with a decrease in production of neurotrophic growth factors, including epidermal growth factor (EGF), nerve growth factor (NGF), insulin-like growth factor (IGF), GDNF (glial cell-derived neurotrophic factor), BDNF (brain-derived neurotrophic factor), and VEGF (vascular endothelial growth factor), which apparently inhibit apoptosis and prompt cell proliferation and differentiation of the developing brain. On the other side, transcription factors comprising JNK (c-Jun N-terminal kinase) and NF-kβ (nuclear factor kappa-beta) also demonstrate an important function in this point [15]. The damage progression proceeds and enters into the tertiary phase by progress in inflammation, alteration in neurogenesis, and impaired synaptogenesis and axonal growth [9].
2.4. Neuronal Death
Neuronal cell death induced by neonatal HI occurs either by necrosis or apoptosis. Necrotic cell death occurs immediately after the insult and is the prevailing pathway of cell death subsequent to irreversible or severe damage [9]. Apoptosis or programmed cell death planned apoptosis is a significant part of ultimate cell death. Apoptosis can last for days and even weeks after the initial insult. It was proposed that, in infants, apoptosis is possibly more critical in provoking cell death compared with necrosis [15].
3. Pharmacological Evidence of Natural Plant Products in Neonatal Hypoxic-Ischemic Brain Damage
3.1. Plant Extracts
3.1.1. Grape Seed Extract
Grape is one of the fruits that is a rich source of phenolic compounds and 60–70% of grape phenolic compounds are discovered in the seeds of the fruit. Grape seed extract (GSE) comprises several phenolic compounds, mainly procyanidins and proanthocyanodins monomers, polymers, and their gallate ester and resveratrol, which is the major compound that is extracted from the skin and seeds of grape [16][17][18]. GSE is a powerful Free Radical Scavenger (FRS) of oxygen and has anti-lipid peroxidation activity [19][20][21][22][23][24][25][16][17][18][26][27][28][29]. GSE revealed anti-oxidant efficacy more than vitamins C and E [30]. GSE also displayed anti-inflammatory and anti-apoptotic actions [31][32][33][34]. In addition, GSE can reduce brain injury in forebrain ischemia in a gerbil model in adults [35].
Zheng and coworkers, in 2005, investigated the neuroprotective effect of GSE against neonatal HI brain injury [36]. In this study, rat pups at postnatal day 7 (P7) were subjected to HI insult and received GSE (50 mg/kg by intra peritoneal (i.p.)) 5 min before hypoxia and 4 h after reoxygenation (twice daily for 1 day). Brain weight loss was reduced from 20.0% in vehicle rats to 3.1% in treated rats, as well as improvement in the histopathologic brain score in hippocampus, thalamus, and cortex after GSE pretreatment. Additionally, lipid peroxidation markers 8-isoPGF2α (8-isoprostaglandin F2α) and TBARS (thiobarbituric acid reactive substances) levels significantly reduced. In another study [37], GSE was injected at 5 min to 5 h after reoxygenation and it was found that GSE post-treatment reduces neurofunctional abnormalities and brain weight loss even when administered 3 h after injury. In addition, the reduction of lipid peroxidation marker 8-isoPGF2α and pro-apoptotic protein c-jun in the rat brain cortex suggested that GSE exhibits neuroprotection through free radical inhibition and anti-apoptotic effects [37].
3.1.2. Grape Seed Proanthocyanidin Extract
Grape seed proanthocyanidin extract (GSPE) is a combination of biologically active flavanol compounds ranging from monomers such as gallic acid, epicatechin, catechins, and their gallate forms, to oligomeric proanthocyanidins. GSPE is known as one of the more potent natural antioxidants and its antioxidant capacity is significantly higher than vitamins E and C [38]. GSPE has been identified in various plants, for instance, cinnamon bark, pine bark, lotus, and apple, and also is widely distributed in red and white grape seeds. Many studies have reported a variety of pharmacological activities for GSPE including antioxidative [39][40], anti-inflammatory, anti-apoptosis [27], and anticancer properties. Recent studies have also been reported that suggest that GSPE possesses neuroprotective effects [40][41][42], for example against an adult rat model of ischemia-reperfusion injury [43].
Tu et al. demonstrated that GSPE significantly decreased brain infarct volume (approximately 50%) and improved neurobehavioral recovery in HIE mice models when pups were pretreated with GSPE (30 mg/kg, intraperitoneal (i.p.) injection) 20 min before HI [44]. In addition, the number of apoptotic cells and pro-apoptotic proteins’ expression (cleaved caspase-3 and bax) was significantly reduced, while expression of anti-apoptosis protein bcl-2 and the bcl2/bax ratio increased in the pretreated group, demonstrating the antiapoptotic role of GSPE [44].
3.2. Phytochemicals
3.2.1. Verbascoside
Phenylethanoid glycosides are a kind of water-soluble natural phenolic compound that have exhibited neuroprotective activity [45]. Verbascoside (VB) (the chemical structure is shown in Figure 1a) is a typical phenylethanoid glycoside from lemon verbena [46]. VB is found in more than 150 types of plants [47]. There is numerous evidence that VB has various biological activities, including antioxidant [48][49], anti-inflammatory [50][51][52], immunoregulatory [53], anticancer [54], and antimicrobial. In addition, several research studies have shown that VB also has neuroprotective properties against various neurological disorders in in vitro and in vivo experiments [55][56][57].
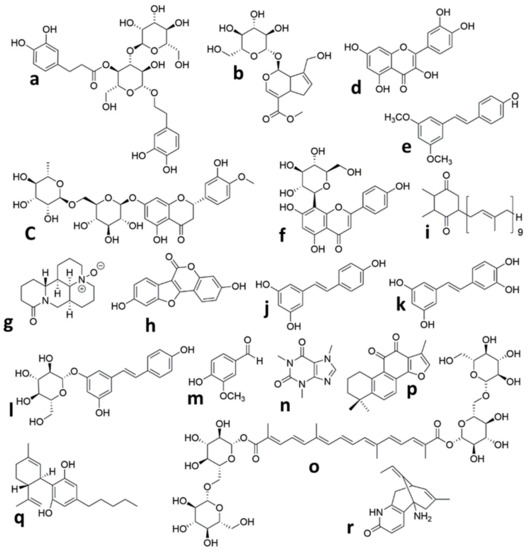
Figure 1. Chemical structures of the isolated compounds from plants in animal models of neonatal Hypoxic Ischemic brain injury. (a) Verbascoside, (b) Geniposide, (c) Hesperidin, (d) Quercetin, (e) Pterostilbene, (f) Vitexin, (g) Oxymatrine, (h) Coumestrol, (i) Plastoquinone, (j) Resveratrol, (k) Piceatannol, (l) Polydatin, (m) Vanillin, (n) Caffeine, (o) Crocin, (p) Tanshinone IIA, (q) Cannabidiol, (r) Huperzine A.
Wei et al. [58] investigated the neuroprotective effects of VB on a HIBD model in rat neonates. In this study, animals were subjected to HIBD at p7 and VB groups (60, 120, and 240 mg/kg) were administered through i.p. injection every 12 h after HI for two consecutive days. VB post-treatment (240 mg/kg) significantly reduced prolonged reflex latencies and brain infarct volume dose-dependently (120 and 240 mg/kg). The VB-treated group (240 mg/kg) also remarkably decreased the degree of degeneration, morphological damage, and necrosis in the cortex and hippocampus CA3 region compared with the HI group. Furthermore, autophagosome formation and the autophagy-related proteins’ expression (P62, Beclin-1, and LC3-II/I ratio) reduced after VB post-treatment.
3.2.2. Geniposide
Iridoid glycosides are phytochemicals that naturally occur in many plants [59][60]. Geniposide (Figure 1b) is one of the major iridoid glycoside compounds that are purified from the Chinese herb Gardenia jasminoides [61]. Nearly 40 plants have been identified that contain geniposide [62]. Geniposide possesses diverse pharmacological activities, including anti-inflammatory [63][64], antidiabetic [65], anti-oxidative, and hepatoprotective [66]. In recent years, geniposide demonstrated excellent neuroprotective activities in experimental neurological dysfunctions, such as Parkinson’s disease [67], Alzheimer’s disease [68][69], and cerebral ischemia [70][71][72].
Liu et al., in 2019, studied the therapeutic effects and underlying mechanisms of geniposide against HI brain damage in neonatal mice. In this study, mice pups were subjected to HI insult at P10 and geniposide was administered (20 mg/kg) daily, intra-gastrically after HI. Findings showed that geniposide treatment significantly reduced the number of apoptotic neurons and also decreased the leakage of serum Immunoglobulin type G (IgG) into brain tissue. In addition, mRNA expression levels of pericyte markers, adherens, and tight junction proteins were upregulated after treatment, which meant that geniposide decreased the disruption of the BBB, which was induced by HI. Moreover, geniposide post-treatment attenuated astrogliosis and microgliosis. Microglia and astrocytes are two of the major neuroinflammation mediators [73]. To further elucidate the underlying molecular mechanism of geniposide-induced neuroprotection, it was found that Phosphoinositide 3-kinases/Protein kinase B (PI3K/Akt) expression signaling pathway-related protein was increased after treatment, which showed that geniposide probably exploits its neuroprotection activity via PI3K/Akt signaling pathway activation.
3.2.3. Hesperidin
Hesperidin (Figure 1c) belongs to the flavanone glycoside class of compounds, which was originally discovered in citrus fruits [74] but has been reported to occur in many plants other than citrus [75]. Several biological activities have been reported from Hesperidin, for example, antioxidative [76][77], anti-inflammatory [78], anticarcinogenic, and antiallergic properties. Hesperidin also has the ability to pass through the BBB [79]. In recent studies, both the in vivo and in vitro neuroprotective properties of hesperidin have been shown [80], which are attributed to its antioxidant and anti-inflammatory activities. This compound also had neuroprotective effects on amyloid β [81] against 3-nitropropionic acid-induced [82][83] and H2O2-induced [84] neurotoxicity. It also possesses antidepressant activities [85]. Another study reported that hesperidin enhances learning and memory in a rat model of cerebral ischemic-reperfusion injury [86].
Rong et al. demonstrated that hesperidin pretreatment in HIBD rat neonates increased the surviving brain volume from 49.8% to 72.9% and improved long-term behavioral development in oral administration of hesperidin (50 mg/kg/day) during 3 days in animals. Hesperidin pretreatment also markedly decreased Fluoro-Jade B stain-positive neurons in the cortex of the brain at post-HI [87]. In the culture media, pretreatment with hesperidin (1.6 μM) reduced the release of LDH (lactate dehydrogenase) and enhanced the levels of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), which indicated that hesperidin promotes neuronal survival. Moreover, levels of ROS and MDA decreased with hesperidin pretreatment in both in vivo and in vitro experimental models. Results also showed that hesperidin pretreatment suppressed FoxO3 phosphorylation, which indicated that possible molecular mechanisms of hesperidin neuroprotection are likely the result of activation of the PI3K/Akt survival signaling pathway and free radical reduction.
References
- Kruszewski, S.P. Neonatal Brain Injury. N. Engl. J. Med. 2005, 352, 1985–1995.
- Lawn, J.E.; Cousens, S.; Zupan, J. 4 Million Neonatal Deaths: When? Where? Why? Lancet 2005, 365, 891–900.
- Dilenge, M.E.; Majnemer, A.; Shevell, M.I. Long-Term Developmental Outcome of Asphyxiated Term Neonates. J. Child Neurol. 2001, 16, 781–792.
- Graham, E.M.; Ruis, K.A.; Hartman, A.L.; Northington, F.J.; Fox, H.E. A Systematic Review of the Role of Intrapartum Hypoxia-Ischemia in the Causation of Neonatal Encephalopathy. Am. J. Obstet. Gynecol. 2008, 199, 587–595.
- Kurinczuk, J.J.; White-Koning, M.; Badawi, N. Epidemiology of Neonatal Encephalopathy and Hypoxic-Ischaemic Encephalopathy. Early Hum. Dev. 2010, 86, 329–338.
- Airede, A.I. Birth Asphyxia and Hypoxic-Ischaemic Encephalopathy: Incidence and Severity. Ann. Trop. Paediatr. 1991, 11, 331–335.
- De Haan, M.; Wyatt, J.S.; Roth, S.; Vargha-Khadem, F.; Gadian, D.; Mishkin, M. Brain and Cognitive-Behavioural Development after Asphyxia at Term Birth. Dev. Sci. 2006, 9, 350–358.
- Roth, S.C.; Edwards, A.D.; Cady, E.B.; Delpy, D.T.; Wyatt, J.S.; Azzopardi, D.; Baudin, J.; Townsend, J.; Stewart, A.L.; Reynolds, E.O. Relation between cerebral oxidative metabolism following birth asphyxia, and neurodevelopmental outcome and brain growth at one year. Dev. Med. Child Neurol. 1992, 34, 285–295.
- Gluckman, P.D.; Pinal, C.S.; Gunn, A.J. Hypoxic-Ischemic Brain Injury in the Newborn: Pathophysiology and Potential Strategies for Intervention. Semin. Neonatol. 2001, 6, 109–120.
- Bunney, P.E.; Zink, A.N.; Holm, A.A.; Billington, C.J.; Kotz, C.M. Orexin activation counteracts decreases in nonexercise activity thermogenesis (NEAT) caused by high-fat diet. Physiol. Behav. 2017, 176, 139–148.
- Gluckman, P.D.; Wyatt, J.S.; Azzopardi, D.; Ballard, R.; Edwards, A.D.; Ferriero, D.M.; Polin, R.A.; Robertson, C.M.; Thoresen, M.; Whitelaw, A.; et al. Selective Head Cooling with Mild Systemic Hypothermia after Neonatal Encephalopathy: Multicentre Randomised Trial. Lancet 2005, 365, 663–670.
- Azzopardi, D.; Strohm, B.; Marlow, N.; Brocklehurst, P.; Deierl, A.; Eddama, O.; Goodwin, J.; Halliday, H.L.; Juszczak, E.; Kapellou, O.; et al. Effects of Hypothermia for Perinatal Asphyxia on Childhood Outcomes. N. Engl. J. Med. 2014, 371, 140–149.
- Shankaran, S.; Pappas, A.; McDonald, S.A.; Vohr, B.R.; Hintz, S.R.; Yolton, K.; Gustafson, K.E.; Leach, T.M.; Green, C.; Bara, R.; et al. Childhood Outcomes after Hypothermia for Neonatal Encephalopathy. Obstet. Gynecol. Surv. 2012, 67, 617–619.
- Ournal, T.H.E.J.; Ediatrics, O.F.P. Neuroprotective Strategies in Neonatal Brain Injury. J. Pediatr. 2017, 192, 22–32.
- Albrecht, M.; Zitta, K.; Groenendaal, F.; Van Bel, F.; Peeters-Scholte, C. Neuroprotective Strategies Following Perinatal Hypoxia-Ischemia: Taking Aim at NOS. Free Radic. Biol. Med. 2019, 142, 123–131.
- Yadav, M.; Jain, S.; Bhardwaj, A.; Nagpal, R.; Puniya, M.; Tomar, R.; Singh, V.; Parkash, O.; Prasad, G.B.K.S.; Marotta, F.; et al. Biological and Medicinal Properties of Grapes and Their Bioactive Constituents: An Update. J. Med. Food 2009, 12, 473–484.
- Jang, M.H.; Piao, X.L.; Kim, J.M.; Kwon, S.W.; Park, J.H. Inhibition of Cholinesterase and Amyloid-β Aggregation by Resveratrol Oligomers from Vitis Amurensis. Phyther. Res. 2008, 22, 544–549.
- Fine, A.M. Oligomeric Proanthocyanidin Complexes: Applications Oligomeric Proanthocyanidins. Altern. Med. Rev. 2000, 5, 144–151.
- Yıldız, E.P.; Ekici, B.; Tatlı, B. Neonatal Hypoxic Ischemic Encephalopathy: An Update on Disease Pathogenesis and Treatment. Expert Rev. Neurother. 2017, 17, 449–459.
- Donega, V.; van Velthoven, C.T.; Nijboer, C.H.; Kavelaars, A.; Heijnen, C.J. The endogenous regenerative capacity of the damaged newborn brain: Boosting neurogenesis with mesenchymal stem cell treatment. J. Cereb. Blood Flow Metab. 2013, 33, 625–634.
- Chen, A.I.; Xiong, L.-J.; Tong, Y.U.; Mao, M. The Neuroprotective Roles of BDNF in Hypoxic Ischemic Brain Injury. Biomed. Rep. 2013, 1, 167–176.
- Yager, J.Y.; Ashwal, S. Animal models of perinatal hypoxic-ischemic brain damage. Pediatr. Neurol. 2009, 40, 156–167.
- Loren, D.J.; Seeram, N.P.; Schulman, R.N.; Holtzman, D.M. Maternal Dietary Supplementation with Pomegranate Juice Is Neuroprotective in an Animal Model of Neonatal Hypoxic-Ischemic Brain Injury. Pediatr. Res. 2005, 57, 858–864.
- Li, X.-L.; Hong, M. Aqueous Extract of Dendrobium Officinale Confers Neuroprotection against Hypoxic-Ischemic Brain Damage in Neonatal Rats. Kaohsiung J. Med. Sci. 2020, 36, 43–53.
- Lafuente, H.; Pazos, M.R.; Alvarez, A.; Mohammed, N.; Santos, M.; Arizti, M.; Alvarez, F.J.; Martinez-Orgado, J.A.; Davidson, J. Effects of Cannabidiol and Hypothermia on Short-Term Brain Damage in New-Born Piglets after Acute Hypoxia-Ischemia. Front. Neurosci. 2016, 10, 1–11.
- Bian, J.T.; Bhargava, H.N. Protective Effects of Grape Seed Proanthocyanidins and Selected Antioxidants against TPA-Induced Hepatic and Brain Lipid Peroxidation and DNA Fragmentation, and Peritoneal Macrophage Activation in Mice. Gen. Pharmacol. 1998, 30, 771–776.
- Bagchi, D.; Sen, C.K.; Ray, S.D.; Das, D.K.; Bagchi, M.; Preuss, H.G.; Vinson, J.A. Molecular Mechanisms of Cardioprotection by a Novel Grape Seed Proanthocyanidin Extract. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2003, 523–524, 87–97.
- Shafiee, M.; Carbonneau, M.A.; Urban, N.; Descomps, B.; Leger, C.L. Grape and Grape Seed Extract Capacities at Protecting LDL against Oxidation Generated by Cu2+, AAPH or SIN-1 and at Decreasing Superoxide THP-1 Cell Production. A Comparison to Other Extracts or Compounds. Free Radic. Res. 2003, 37, 573–584.
- Balu, M.; Sangeetha, P.; Murali, G.; Panneerselvam, C. Modulatory Role of Grape Seed Extract on Age-Related Oxidative DNA Damage in Central Nervous System of Rats. Brain Res. Bull. 2006, 68, 469–473.
- Ariga, T. The Antioxidative Function, Preventive Action on Disease and Utilization of Proanthocyanidins. BioFactors 2004, 21, 197–201.
- Sato, M.; Bagchi, D.; Tosaki, A.; Das, D.K. Grape Seed Proanthocyanidin Reduces Cardiomyocyte Apoptosis by Inhibiting Ischemia/Reperfusion-Induced Activation of JNK-1 and C-JUN. Free Radic. Biol. Med. 2001, 31, 729–737.
- Ray, S.D.; Kumar, M.A.; Bagchi, D. A novel proanthocyanidin IH636 grape seed extract increases in vivo Bcl-XL expression and prevents acetaminophen-induced programmed and unprogrammed cell death in mouse liver. Arch. Biochem. Biophys. 1999, 369, 42–58.
- Sugisawa, A.; Inoue, S.; Umegaki, K. Grape Seed Extract Prevents H2O2-Induced Chromosomal Damage in Human Lymphoblastoid Cells. Biol. Pharm. Bull. 2004, 27, 1459–1461.
- Baines, C.P.; Molkentin, J.D. STRESS signaling pathways that modulate cardiac myocyte apoptosis. J. Mol. Cell. Cardiol. 2005, 38, 47–62.
- Hwang, I.K.; Yoo, K.Y.; Kim, D.S.; Jeong, Y.K.; Kim, J.D.; Shin, H.K.; Lim, S.S.; Yoo, I.D.; Kang, T.C.; Kim, D.W.; et al. Neuroprotective Effects of Grape Seed Extract on Neuronal Injury by Inhibiting DNA Damage in the Gerbil Hippocampus after Transient Forebrain Ischemia. Life Sci. 2004, 75, 1989–2001.
- Feng, Y.; Liu, Y.M.; Fratkins, J.D.; LeBlanc, M.H. Grape Seed Extract Suppresses Lipid Peroxidation and Reduces Hypoxic Ischemic Brain Injury in Neonatal Rats. Brain Res. Bull. 2005, 66, 120–127.
- Feng, Y.; Liu, Y.M.; Leblanc, M.H.; Bhatt, A.J.; Rhodes, P.G. Grape Seed Extract given Three Hours after Injury Suppresses Lipid Peroxidation and Reduces Hypoxic-Ischemic Brain Injury in Neonatal Rats. Pediatr. Res. 2007, 61, 295–300.
- Fracassetti, D.; Costa, C.; Moulay, L.; Tomás-barberán, F.A. Ellagic Acid Derivatives, Ellagitannins, Proanthocyanidins and Other Phenolics, Vitamin C and Antioxidant Capacity of Two Powder Products from Camu-Camu Fruit (Myrciaria Dubia). Food Chem. 2013, 139, 578–588.
- Margalef, M.; Guerrero, L.; Pons, Z.; Isabel, F.; Arola, L.; Muguerza, B.; Arola-arnal, A. A dose—Response study of the bioavailability of grape seed proanthocyanidin in rat and lipid-lowering effects of generated metabolites in HepG2 cells. Food Res. Int. 2014, 64, 500–507.
- Devi, S.A.; Jolitha, A.B.; Ishii, N. Grape seed proanthocyanidin extract (GSPE) and antioxidant defense in the brain of adult rats. Med Sci. Monit. 2006, 12, BR124–BR129.
- Ferruzzi, M.G.; Lobo, J.K.; Janle, E.M.; Cooper, B.; Simon, J.E.; Wu, Q.; Welch, C.; Ho, L.; Weaver, C.; Pasinetti, G.M. Bioavailability of gallic acid and catechins from grape seed polyphenol extract is improved by repeated dosing in rats: Implications for treatment in alzheimer’s disease. J. Alzheimers Dis. 2009, 18, 113–124.
- Wu, L.; Zhang, Q.L.; Zhang, X.Y.; Lv, C.; Li, J.; Yuan, Y.; Yin, F.X. Pharmacokinetics and Blood-Brain Barrier Penetration of (+)-Catechin and (-)-Epicatechin in Rats by Microdialysis Sampling Coupled to High-Performance Liquid Chromatography with Chemiluminescence Detection. J. Agric. Food Chem. 2012, 60, 9377–9383.
- Kong, X.; Guan, J.; Gong, S.; Wang, R. Neuroprotective Effects of Grape Seed Procyanidin Extract on Ischemia-Reperfusion Brain Injury. Chin. Med. Sci. J. 2017, 32, 92–99.
- Tu, X.; Wang, M.; Liu, Y.; Zhao, W.; Ren, X.; Li, Y.; Liu, H.; Gu, Z.; Jia, H.; Liu, J.; et al. Pretreatment of Grape Seed Proanthocyanidin Extract Exerts Neuroprotective Effect in Murine Model of Neonatal Hypoxic-Ischemic Brain Injury by Its Antiapoptotic Property. Cell. Mol. Neurobiol. 2019, 39, 953–961.
- Koo, K.A.; Sung, S.H.; Park, J.H.; Kim, S.H.; Lee, K.Y.; Kim, Y.C. In Vitro Neuroprotective Activities of Phenylethanoid Glycosides from Callicarpa Dichotoma. Planta Med. 2005, 71, 778–780.
- Alipieva, K.; Korkina, L.; Erdogan, I.; Georgiev, M.I. Verbascoside—A review of its occurrence, (bio) synthesis and pharmacological significance. Biotechnol. Adv. 2014, 32, 1065–1076.
- Taylor, P.; He, J.; Hu, X.; Zeng, Y.; Li, Y.; Wu, H.; Qiu, R.; Ma, W.; Li, T.; Li, C.; et al. Advanced Research on Acteoside for Chemistry and Bioactivities. J. Asian Nat. Prod. Res. 2011, 37–41.
- Imperio, M.D.; Cardinali, A.; Antuono, I.D.; Linsalata, V.; Minervini, F.; Redan, B.W.; Ferruzzi, M.G. Stability—Activity of Verbascoside, a Known Antioxidant Compound, at Different PH Conditions. Food Res. Int. 2014, 66, 373–378.
- Frum, Y.; Viljoen, A.M.; Van Heerden, F.R. Verbascoside and luteolin-5-o-β-d-glucoside isolated from Halleria lucida L. exhibit antagonistic anti-oxidant properties in vitro. S. Afr. J. Bot. 2007, 73, 583–587.
- Lee, J.H.; Lee, J.Y.; Kang, H.S.; Jeong, C.H.; Moon, H.; Whang, W.K.; Kim, C.J.; Sim, S.S. The Effect of Acteoside on Histamine Release and Arachidonic Acid Release in RBL-2H3 Mast Cells. Arch. Pharmacal Res. 2006, 29, 508–513.
- Speranza, L.; Franceschelli, S.; Pesce, M.; Reale, M.; Menghini, L.; Vinciguerra, I.; De Lutiis, M.A.; Felaco, M.; Grilli, A. Antiinfl Ammatory Effects in THP-1 Cells Treated with Verbascoside. Phytother. Res. 2010, 1404, 1398–1404.
- Pesce, M.; Franceschelli, S.; Ferrone, A.; Anna, M.; Lutiis, D.; Patruno, A.; Grilli, A.; Felaco, M.; Speranza, L. Verbascoside Down-Regulates Some pro-Inflammatory Signal Transduction Pathways by Increasing the Activity of Tyrosine Phosphatase SHP-1 in the U937 Cell Line. J. Cell. Mol. Med. 2015, 19, 1548–1556.
- Hno, T.O.; Noue, M.I.; Gihara, Y.O.; Aracoglu, I.S. Antimetastatic Activity of Acteoside, a Phenylethanoid Glycoside. Biol. Pharm. Bull. 2002, 25, 666–668.
- Zhou, L.; Feng, Y.; Jin, Y.; Liu, X.; Sui, H.; Chai, N.; Chen, X.; Liu, N.; Ji, Q.; Wang, Y.; et al. Verbascoside Promotes Apoptosis by Regulating HIPK2-P53 Signaling in Human Colorectal Cancer. BMC Cancer 2014, 14, 1–11.
- Li, N.; Wang, J.; Ma, J.; Gu, Z.; Jiang, C.; Yu, L.; Fu, X. Neuroprotective Effects of Cistanches Herba Therapy on Patients with Moderate Alzheimer ’ s Disease. Evid. Based Complement. Altern. Med. 2015, 2015, 103985.
- Wu, J.; Huang, F.X.; Wang, J.; Shi, C.C.; Fang, G.Y. Protective Effect of Liquiritin on Corticosterone-Induced Neurotoxicity in Pc12 Cells. Trop. J. Pharm. Res. 2018, 17, 2013–2017.
- Yuan, J.; Ren, J.; Wang, Y.; He, X.; Zhao, Y. Acteoside Binds to Caspase-3 and Exerts Neuroprotection in the Rotenone Rat Model of Parkinson’s Disease. PLoS ONE 2016, 11, e0162696.
- Wei, W.; Lu, M.; Lan, X.; Liu, N.; Wang, H.; Du, J.; Sun, T.; Li, Y.; Yu, J. Neuroprotective Effect of Verbascoside on Hypoxic-Ischemic Brain Damage in Neonatal Rat. Neurosci. Lett. 2019, 711, 134415.
- Inda, B.D.; Ebnath, S.D.; Anik, R.B. Naturally Occurring Iridoids and Secoiridoids. An Updated Review, Part 4. Chem. Pharm. Bull. 2011, 59, 803–833.
- El-Naggar, L.J.; Beal, J.L. Iridoids. A review. J. Nat. Prod. 1980, 43, 649–707.
- Chou, G.; Wang, Z.; Medica, C.M.; Park, Z.H. Iridoid glycosides from Gardenia jasminoides Ellis. Helv. Chim. Acta 2008, 91, 4–10.
- Shan, M.; Yu, S.; Yan, H.; Guo, S.; Xiao, W.; Wang, Z.; Zhang, L.; Ding, A.; Wu, Q.; Fong, S.; et al. A review on the phytochemistry, pharmacology, pharmacokinetics and toxicology of geniposide, a natural product. Molecules 2017, 22, 1689.
- Li, F.; Li, W.; Li, X.; Li, F.; Zhang, L.; Wang, B.; Huang, G.; Guo, X.; Wan, L.; Liu, Y.; et al. Geniposide Attenuates Inflammatory Response by Suppressing P2Y14 Receptor and Downstream ERK1/2 Signaling Pathway in Oxygen and Glucose Deprivation-Induced Brain Microvascular Endothelial Cells. J. Ethnopharmacol. 2016, 185, 77–86.
- Wang, J.; Hou, J.; Zhang, P. Geniposide Reduces Inflammatory Responses of Oxygen-Glucose Deprived Rat Microglial Cells via Inhibition of the TLR4 Signaling Pathway. Neurochem. Res. 2012, 2, 2235–22480.
- Guo, L.; Xia, Z.; Gao, X.; Yin, F.; Liu, J. Glucagon-like Peptide 1 Receptor Plays a Critical Role in Geniposide-Regulated Insulin Secretion in INS-1 Cells. Acta Pharmacol. Sin. 2011, 33, 237–241.
- Liu, J.; Yin, F.; Zheng, X.; Jing, J.; Hu, Y. Geniposide, a Novel Agonist for GLP-1 Receptor, Prevents PC12 Cells from Oxidative Damage via MAP Kinase Pathway. Neurochem. Int. 2007, 51, 361–369.
- Chen, Y.; Zhang, Y.; Li, L.; Hölscher, C. Neuroprotective Effects of Geniposide in the MPTP Mouse Model of Parkinson’s Disease. Eur. J. Pharmacol. 2015, 768, 21–27.
- Zhang, H.; Zhao, C.; Lv, C.; Liu, X.; Du, S.; Li, Z.; Wang, Y.; Zhang, W. Geniposide Alleviates Amyloid-Induced Synaptic Injury by Protecting Axonal Mitochondrial Trafficking. Front. Cell. Neurosci. 2017, 10, 1–13.
- Lv, C.; Wang, L.; Liu, X.; Yan, S.; Yan, S.S.; Wang, Y.; Zhang, W. Multi-Faced Neuroprotective Effects of Geniposide Depending on the RAGE-Mediated Signaling in an Alzheimer Mouse Model. Neuropharmacology 2015, 89, 175–184.
- Wang, J.; Li, D.; Hou, J.; Lei, H. Protective Effects of Geniposide and Ginsenoside Rg1 Combination Treatment on Rats Following Cerebral Ischemia Are Mediated via Microglial MicroRNA-155-5p Inhibition. Mol. Med. Rep. 2018, 17, 3186–3193.
- Pan, L.; Wang, W.; Shi, F.; Zhou, J.; Zhang, M.; Zhu, H.; Zeng, M. Exploratory Pharmacokinetics of Geniposide in Rat Model of Cerebral Ischemia Orally Administered with or without Baicalin and/or Berberine. Evid. Based Complement. Altern. Med. 2013, 2013, 349531.
- Huang, B.; Chen, P.; Huang, L.; Li, S.; Zhu, R.; Sheng, T.; Yu, W.; Chen, Z.; Wang, T. Geniposide Attenuates Post-Ischaemic Neurovascular Damage via GluN2A/AKT/ ERK-Dependent Mechanism. Cell. Physiol. Biochem. 2017, 43, 705–716.
- Liu, F.; Wang, Y.; Yao, W.; Xue, Y.; Zhou, J.; Liu, Z. Geniposide Attenuates Neonatal Mouse Brain Injury after Hypoxic-Ischemia Involving the Activation of PI3K/Akt Signaling Pathway. J. Chem. Neuroanat. 2019, 102, 54.
- Wu, G.A.; Terol, J.; Ibanez, V.; López-García, A.; Pérez-Román, E.; Borredá, C.; Domingo, C.; Tadeo, F.R.; Carbonell-caballero, J.; Alonso, R.; et al. Genomics of the Origin and Evolution of Citrus. Nature 2018.
- Garg, A.; Garg, S.; Zaneveld, L.J.D.; Singla, A.K. Chemistry and Pharmacology of the Citrus Bioflavonoid Hesperidin. Phyther. Res. 2001, 15, 655–669.
- Elavarasan, J.; Velusamy, P.; Ganesan, T.; Ramakrishnan, S.K.; Rajasekaran, D.; Periandavan, K. Hesperidin-Mediated Expression of Nrf2 and Upregulation of Antioxidant Status in Senescent Rat Heart. J. Pharm. Pharmacol. 2012, 64, 1472–1482.
- Cho, J. Antioxidant and Neuroprotective Effects of Hesperidin and Its Aglycone Hesperetin. Arch. Pharmacal Res. 2006, 29, 699–706.
- Lorzadeh, E.; Ramezani-Jolfaie, N.; Mohammadi, M. The Effect of Hesperidin Supplementation on Inflammatory Markers in Human Adults: A Systematic Review and Meta-Analysis of Randomized Controlled Clinical Trials. Chem. Biol. Interact. 2019, 307, 8–15.
- Dimpfel, W. Different Anticonvulsive Effects of Hesperidin and Its Aglycone Hesperetin on Electrical Activity in the Rat Hippocampus In-Vitro. J. Pharm. Pharmacol. 2006, 58, 375–379.
- Rainey-smith, S.; Schroetke, L.; Bahia, P.; Fahmi, A.; Skilton, R.; Spencer, J.P.E.; Rice-evans, C.; Rattray, M.; Williams, R.J. Neuroprotective effects of hesperetin in mouse primary neurones are independent of CREB activation. Neurosci. Lett. 2008, 438, 29–33.
- Huang, S.; Tsai, S.; Lin, J.; Wu, C.; Yen, G. Cytoprotective Effects of Hesperetin and Hesperidin against Amyloid β-Induced Impairment of Glucose Transport through Downregulation of Neuronal Autophagy. Mol. Nutr. Food Res. 2012, 601–609.
- Menze, E.T.; Tadros, M.G.; Abdel-tawab, A.M.; Khalifa, A.E. Potential Neuroprotective Effects of Hesperidin on 3-Nitropropionic Acid-Induced Neurotoxicity in Rats. Neurotoxicology 2012, 33, 1265–1275.
- Kumar, P.; Kumar, A. Protective Effect of Hesperidin and Naringin against 3-Nitropropionic Acid Induced Huntington’s like Symptoms in Rats: Possible Role of Nitric Oxide. Behav. Brain Res. 2010, 206, 38–46.
- Choi, E.J.; Ahn, W.S. Neuroprotective Effects of Chronic Hesperetin Administration in Mice. Arch. Pharmacal Res. 2008, 31, 1457–1462.
- Donato, F.; de Gomes, M.G.; Goes, A.T.R.; Borges Filho, C.; Del Fabbro, L.; Antunes, M.S.; Souza, L.C.; Boeira, S.P.; Jesseb, C.R. Hesperidin Exerts Antidepressant-like Effects in Acute and Chronic Treatments in Mice: Possible Role of l-Arginine-NO-CGMP Pathway and BDNF Levels. Brain Res. Bull. 2014, 104, 19–26.
- Gaur, V.; Kumar, A. Hesperidin Pre-Treatment Attenuates NO-Mediated Cerebral Ischemic Reperfusion Injury and Memory Dysfunction. Pharmacol. Rep. 2010, 62, 635–648.
- Rong, Z.; Pan, R.; Xu, Y.; Zhang, C.; Cao, Y.; Liu, D. Hesperidin pretreatment protects hypoxia—Ischemic brain injury in neonatal rat. Neuroscience 2013, 255, 292–299.


