 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Carmelo Pozzo | + 1848 word(s) | 1848 | 2020-10-21 05:43:29 | | | |
| 2 | Rita Xu | -724 word(s) | 1124 | 2021-01-19 04:31:42 | | |
Video Upload Options
Low muscle mass is significantly associated with low tolerability of chemotherapy and poor survival. In this context, despite the great strides in the treatment of cancer, targeted therapies such as tyrosine kinase inhibitors (TKIs) could exacerbate loss of muscle mass in cancer patients. In the last decades, the impact of muscle mass wasting during TKI treatment on clinical outcomes has been put under the spotlights. We evaluated different molecular pathways of TKIs potentially involved in muscle wasting assessing the effects of different TKI therapies – such as sorafenib, regorafenib, sunitinib, lenvatinib - on muscle mass, and highlighted their potential clinical implications. The assessment of muscle mass from CT imaging could be helpful in predicting toxicity and prognosis in patients treated with TKI such as sorafenib. An early recognition of low muscle mass and an effective personalized nutritional support could prevent or attenuate muscle mass wasting. However, the role of nutrition is still overlooked and future clinical trials are needed to find the optimal nutritional support to countermeasure muscle mass depletion during TKI therapy.
1. Introduction
The discovery of the overexpression of kinases in various cancers has led to the development of tyrosine kinase inhibitors (TKIs). TKIs demonstrated to produce a significant improvement in survival rates in several cancers. Just to name a few examples, imatinib has revolutionized the treatment of chronic myelogenous leukemia as well as gastrointestinal stromal tumors (GISTs) [1]; sorafenib was the first therapy proven to prolong survival in patients with metastatic hepatocellular carcinoma (HCC) [2]; sunitinib provided for the first time a survival advantage over interferon to treat metastatic renal cell cancer (RCC) [3]. However, despite the great strides in the treatment of cancer, these agents can also induce frustrating dose-limiting toxicities (DLTs) partially due to the loss of body weight and muscle mass [4]. Indeed, the reduction in physical activity, the nutritional deficiencies resulting from cancer itself, and the side effects of oncologic treatment—such as nausea, vomiting, diarrhea, taste alteration, and early satiety—often lead to a loss of weight and lean tissue mass. Patients with poor nutritional status are often not able to tolerate chemotherapy and discontinue treatment. Low skeletal muscle mass was already shown to be a significant predictor of chemotherapy toxicity and survival in cancer patients [5][6][7]. Thus, research increasingly focuses on the importance of preserving skeletal muscle mass of patients receiving chemotherapy. In recent decades, a growing number of studies attempted to understand whether muscle wasting was exacerbated by TKI treatment and if such muscle loss was associated with toxicity and survival outcomes.
2. Molecular Pathways of TKI Involved in Muscle Wasting
The maintenance of skeletal muscle mass is determined by a close balance between protein synthesis and protein degradation. Intracellular signaling cascades, regulating the mechanisms for muscle growth or muscle loss, initiate with a variety of chemical signals depending on nutritional and hormonal status—such as insulin or insulin-like growth factor-1 (IGF-1)—or energy state (AMP kinase) and physical activity, or other mediators of environmental stress (glucocorticoids, cytokines). A key point of integration in muscle protein synthesis is the phosphoinositide 3-kinase (PI3K)/thymoma viral proto-oncogene (AKT) kinase [8]. The insulin/IGF-1- Akt pathway increases skeletal muscle protein synthesis via inhibiting glycogen synthase kinase 3β and activating the mechanistic target of rapamycin complex 1 (mTORC1) signaling [9]. In turn, mTORC1 activates translation initiation and elongation, and ribosome biogenesis of proteins, and consequently muscle cell growth [10].
Tyrosine kinases are enzymes that target proteins involved in diverse normal cellular regulatory processes [11]. The receptors of tyrosine kinases are membrane-spanning cell surface proteins linking extracellular signals to the cytoplasm [12]. Ligand binding induces dimerization of these receptors, resulting in autophosphorylation of their cytoplasmic domains and activation of tyrosine kinase activity [11]. TKIs target different receptor tyrosine kinases such as vascular endothelial growth factor receptors (VEGFRs). VEGFRs 1/2/3 are located on vascular endothelial cells and play a key role in angiogenesis [13]. In healthy adults, angiogenesis is a complex multistep process that is tightly controlled by a balance between endogenous pro-angiogenic and anti-angiogenic factors. However, angiogenesis is a crucial prognostic factor in cancer and frequently correlates with tumor progression, disease severity, and metastatic potential [14]. TKIs such as sorafenib and sunitinib partly exert their anti-tumor activity by inhibiting the tyrosine kinase activity of VEGFR-2 [15]. Platelet-derived growth factor receptors (PDGFRs) are also TKIs’ targets involved in angiogenesis: mutational activation or upregulation of PDGFRs lead to uncontrolled blood vessel formation and cancer. Moreover, PDGFR-β emerged as a key regulator of cell growth and division and mediates a significant impact on malignant cells and tumor microenvironment [16]. Epidermal growth factor receptors (EGFRs) are transmembrane protein receptors for extracellular protein ligands belonging to the group of epidermal growth factor (EGF) [17]. The overexpression of the EGFR is associated with the development of a wide variety of tumors such as non-small cell lung cancer and breast cancer [18]. Finally, KIT, RET, and B-RAF are other receptor tyrosine kinases encoded by proto-oncogenes: the overexpression or mutations of these proteins can promote carcinogenesis in several tissues [19].
All these tyrosine kinase receptors have been associated with the signaling pathway of activation of the PI3K/AKT/mTOR, which promotes cell growth, survival, and proliferation. In many cancers, the PI3K/AKT/mTOR pathway is overactive, thus reducing apoptosis and allowing cell proliferation [20]. On the other hand, the activation of the AKT/mTOR pathway and its downstream targets is essential for regulating skeletal muscle fiber size [21]. In vivo, genetic activation of the AKT/mTOR pathway caused muscle hypertrophy and prevented atrophy, whereas blocking of this pathway blocked muscle hypertrophy [22]. By inhibiting receptor tyrosine kinase signaling, TKIs have shown to indirectly suppress AKT and mTOR. In particular, sorafenib blocks VEGFR, PDGFR, c-KIT, and RET and inhibits downstream Raf serine/threonine kinase activity to prevent tumor growth, as demonstrated in advanced HCC, RCC, and unresectable thyroid cancer [23]. Additionally, regorafenib is an oral multikinase inhibitor of VEGFR1-3, KIT, RET, B-RAF, and PDGFR [24]. As illustrated in HCC in Figure 1, several TKIs indirectly inhibit mTOR and consequently impair cell proliferation, protein synthesis, and muscle growth.
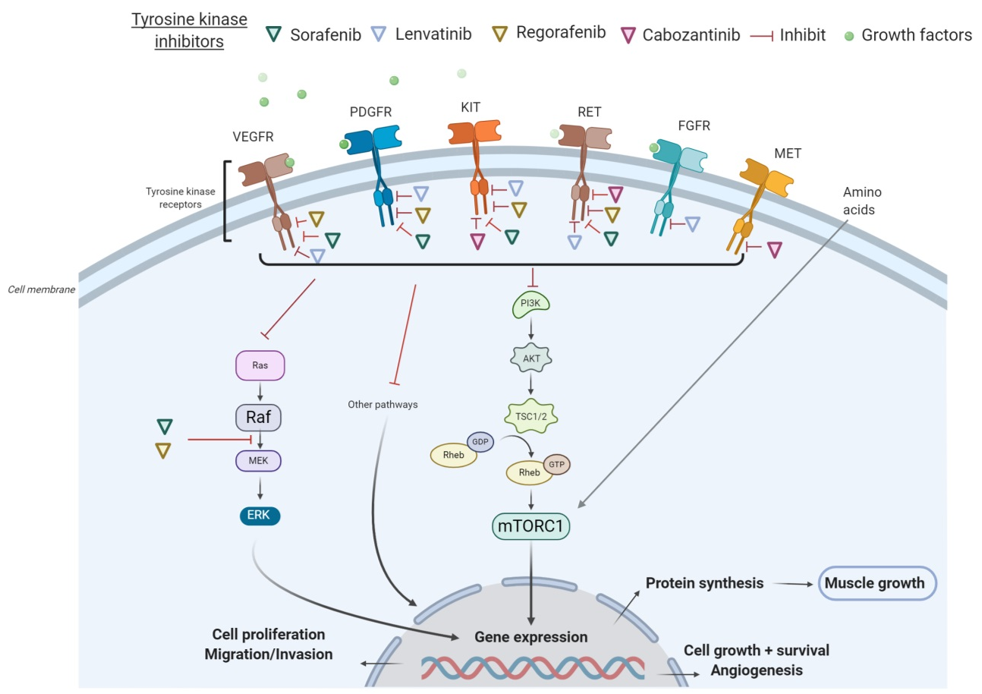
Figure 1. Mechanisms of action of tyrosine kinase inhibitors (TKIs) in hepatocellular carcinoma (HCC). Abbreviations: FGFR, fibroblast growth factor receptors; GDP, guanosine diphosphate; GTP, guanosine triphosphate; mTORC1, mammalian target of rapamycin complex 1; PDGFR, platelet-derived growth factor receptor; PI3K, phosphoinositide 3-kinase; TSC, tuberous sclerosis complex; VEGFR, vascular endothelial growth factor receptor.
Although the effects of mTOR inhibitors on muscle mass have still to be fully elucidated, a recent human study [25] demonstrated that the long-term use of mTOR inhibitors induces a marked loss of muscle mass. This confirms that TKIs may alter muscle mass, probably due to interferences with pathways of AKT/mTOR. As reported in the next paragraph, several studies assessed the impact of TKI treatment on muscle mass in cancer patients.
References
- Dagher, ; Cohen, M.; Williams, G.; Rothmann, M.; Gobburu, J.; Robbie, G.; Rahman, A.; Chen, G.; Staten, A.; Griebel, D.; et al. Approval summary: Imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin. Cancer Res. 2002, 8, 3034–3038.
- Llovet, M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N Engl. J. Med. 2008, 359, 378–390.
- Coppin, Sunitinib for advanced renal cell cancer. Biology 2008, 2, 97–105.
- Mir, ; Coriat, R.; Blanchet, B.; Durand, J.P.; Boudou-Rouquette, P.; Michels, J.; Ropert, S.; Vidal, M.; Pol, S.; Chaussade, S.; et al. Sarcopenia predicts early dose-limiting toxicities and pharmacokinetics of sorafenib in patients with hepatocellular carcinoma. PLoS ONE 2012, 7, e37563, doi:10.1371/journal.pone.0037563.
- Rinninella, ; Cintoni, M.; Raoul, P.; Pozzo, C.; Strippoli, A.; Bria, E.; Tortora, G.; Gasbarrini, A.; Mele, M.C. Muscle mass, assessed at diagnosis by L3-CT scan as a prognostic marker of clinical outcomes in patients with gastric cancer: A systematic review and meta-analysis. Clin. Nutr. 2020, 39, 2045–2054.
- Rinninella, ; Fagotti, A.; Cintoni, M.; Raoul, P.; Scaletta, G.; Scambia, G.; Gasbarrini, A.; Mele, M.C. Skeletal muscle mass as a prognostic indicator of outcomes in ovarian cancer: A systematic review and meta-analysis. Int. J. Gynecol Cancer 2020, 30, 654–663.
- Shachar, S.; Williams, G.R.; Muss, H.B.; Nishijima, T.F. Prognostic value of sarcopenia in adults with solid tumours: A meta-analysis and systematic review. Eur. J. Cancer 2016, 57, 58–67.
- Glass, J. Pi3 kinase regulation of skeletal muscle hypertrophy and atrophy. Curr. Top. Microbiol. Immunol. 2010, 346, 267–278.
- Schiaffino, ; Mammucari, C. Regulation of skeletal muscle growth by the IGF1- Akt/PKB pathway: Insights from genetic models. Skelet. Muscle. 2011, 1, 4.
- Adegoke, A.; Abdullahi, A.; Tavajohi-Fini, P. mTORC1 and the regulation of skeletal muscle anabolism and mass. Appl. Physiol. Nutr. Metab. 2012, 37, 395–406.
- Arora, ; Scholar, E.M. Role of tyrosine kinase inhibitors in cancer therapy. J. Pharm. Exp. 2005, 315, 971–979, doi:10.1124/jpet.105.084145.
- Pawson, Regulation and targets of receptor tyrosine kinases. Eur. J. Cancer 2002, 38, S3–S10.
- Shibuya, Vascular endothelial growth factor (VEGFR) and its receptor (VEGFR) signaling in angiogenesis: A crucial target for anti- and pro-angiogenic therapies. Genes Cancer 2011, 2, 1097–1105.
- Mendel, B.; Laird, A.D.; Smolich, B.D.; Blake, R.A.; Liang, C.; Hannah, A.L.; Shaheen, R.M.; Ellis, L.M.; Weitman, S.; Shawver, L.K.; et al. Development of SU5416, a selective small molecule inhibitor of VEGF receptor tyrosine kinase activity, as an anti-angiogenesis agent. Anticancer Drug Des. 2000, 15, 29–41.
- Gotink, J.; Verheul, H.M. Anti-angiogenic tyrosine kinase inhibitors: What is their mechanism of action? Angiogenesis 2010, 13, 1–14.
- Nordby, ; Richardsen, E.; Rakaee, M.; Ness, N.; Donnem, T.; Patel, H.R.; Busund, L.T.; Bremnes, R.M.; Andersen, S. High expression of PDGFR-β in prostate cancer stroma is independently associated with clinical and biochemical prostate cancer recurrence. Sci. Rep. 2017, 7, 43378.
- Wieduwilt, J.; Moasser, M.M. The epidermal growth factor receptor family: Biology driving targeted therapeutics. Cell Mol. Life Sci. 2008, 65, 1566–1584.
- Sigismund, ; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20.
- Du, ; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58.
- Xu, ; Na, L.; Li, Y.; Chen, L. Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci. 2020, 10, 54.
- Bodine, C.; Stitt, T.N.; Gonzalez, M.; Kline, W.O.; Stover, G.L.; Bauerlein, R.; Zlotchenko, E.; Scrimgeour, A.; Lawrence, J.C.; Glass, D.J.; et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol. 2001, 3, 1014–1019.
- Edinger, L.; Thompson, C.B. Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol. Biol. Cell. 2002, 13, 2276–2288.
- Adnane, ; Trail, P.A.; Taylor, I.; Wilhelm, S.M. Sorafenib (BAY 43-9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzym. 2006, 407, 597–612.
- Wilhelm, M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schütz, G.; Thierauch, K.H.; Zopf, D. Regorafenib (BAY 73-4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 2011, 129, 245–255.
- Gyawali, B.; Shimokata, T.; Honda, K.; Kondoh, C.; Hayashi, N.; Yoshino, Y.; Sassa, N.; Nakano, Y.; Gotoh, M.; Ando, Y. Muscle wasting associated with the long-term use of mTOR inhibitors. Mol. Clin. Oncol. 2016, 5, 641–646.


