 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Helena Ferreira | + 4984 word(s) | 4984 | 2021-01-06 08:57:31 | | | |
| 2 | Rita Xu | -1800 word(s) | 3184 | 2021-01-15 07:47:31 | | |
Video Upload Options
Rheumatoid arthritis (RA) is a highly debilitating chronic inflammatory autoimmune disease most prevalent in women. The true etiology of this disease is complex, multifactorial, and is yet to be completely elucidated. Changes in the lipid profile at a molecular level in RA are still poorly understood. Studies on the variation of lipid profile in RA using lipidomics showed that fatty acid and phospholipid profile, especially in phosphatidylcholine and phosphatidylethanolamine, are affected in this disease. These promising results could lead to the discovery of new diagnostic lipid biomarkers for early diagnosis of RA and targets for personalized medicine.
1. Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a chronic inflammatory autoimmune disease (AID) affecting almost 1% of the population worldwide and is most prevalent in women [1]. In RA, chronic inflammation of the synovium membrane is a significant pathologic feature, leading to progressive and irreversible joint destruction, deformity, and disability, if left untreated [2]. Increased autoantibody production is another hallmark of RA, in particular, the rheumatoid factor (RF) and the anticitrullinated protein antibodies (ACPA) [3]. Both RF and ACPA have predictive, diagnostic, and prognostic roles, since it is possible to detect these autoantibodies prior to RA onset and their presence positively correlates with disease severity and joint destruction [4][5]. However, only about 70% of RA patients produce RF or ACPA [6], thus revealing that the diagnosis of RA cannot be based solely on these biomarkers.
Besides the presence of autoantibodies, the etiology of RA is complex, multifactorial, and is yet to be completely elucidated. The main physiological characteristics of RA (joint inflammation and cartilage destruction) are the result of the infiltration of immune cells into the synovial joint lining and the associated complex network of cytokines [3][7][8]. Impaired adaptive immune responses are considered keys in RA pathogenesis. In brief, activated T helper cells release several pro-inflammatory cytokines into the synovial membrane and fluid [9]. Another set of pro-inflammatory cytokines is secreted by recently activated macrophages contributing to a self-amplifying pro-inflammatory loop. This induces the proliferation and differentiation of B cells, resulting in the production of RF and ACPA autoantibodies [3]. Fibroblasts from the synovial membrane produce matrix metalloproteinases that will destroy the cartilage and activate osteoclasts, promoting bone resorption. The release of such pro-inflammatory mediators induces protein modifications that enhance the immune response and chronic inflammation [10][11].
Additional pivotal cells for RA pathogenesis are monocytes and neutrophils. It has been suggested that an increased turnover of monocytes, migrating from the bone marrow into the inflamed synovia, occurs in RA [12]. The process of recruiting neutrophils into the synovium is a key feature of inflammatory responses in RA, and it is propelled, once more, by an intricate network of cytokines [3]. The aforementioned pro-inflammatory cytokines released by macrophages and T cells serve as a primer of the infiltrated neutrophils. Once primed, neutrophils will foster joint destruction by inducing synthesis of proteins that can upregulate and extend neutrophil ability to secrete a large quantity of cytokines and chemokines [13][14]. Furthermore, neutrophils contribute to cartilage destruction in the synovial fluid, damage surrounding tissues, induce oxidative stress conditions due to the release of reactive oxygen species (ROS), increasing the inflammation status, and may be a source of ACPA contributing for the impaired immune response in RA [15][16][17].
Given the complex character of RA, it is believed that genetic and environmental factors also take part in the pathogenesis of this disease. RA susceptibility is strongly associated with major histocompatibility complex genes, and the genetic factors have a straight connection with the environmental factors [18]. The production of ACPA may be triggered by silica exposure, microbiota, infections, epigenetic modifications and environmental risk factors, especially cigarette smoking [19]. The microbiota of RA patients differs from that of the general population and is associated with the inflammatory conditions of this disease, while viral and bacterial infections contribute for the development of the autoimmune response in RA [20]. Additionally, RA has an increased heritability that is estimated to be 50% for ACPA-seronegative and 70% for ACPA-seropositive [21][22]. The expression of ACPA in RA patients is associated with the presence of shared epitope alleles (SE) which, in turn, are associated with disease severity [23]. In genetically predisposed individuals, the environmental factors have a high impact on the risk of developing RA by inducing molecular changes to proteins that trigger a loss of immunological self-tolerance [3].
Systemic effects linked to inflammation are responsible for severe physical disability of RA patients, diminishing their life quality. Impaired muscle function may be considered a hallmark of RA and several factors contribute to this process. Pro-inflammatory cytokines, as mentioned before, are highly involved in RA pathogenesis. Cytokines are responsible for low skeletal muscle function in RA patients by reducing muscle fibers contractility due to increased oxidant activity in the system [24]. Cytokines were found to be two times higher in RA patients’ muscle compared with systemic cytokines levels [25]. The chronic inflammation in RA is also involved in skeletal muscle pathology. Inflammation and insulin resistance are correlated with diminished mitochondrial function and content, thus disrupting muscle oxidative metabolic capacity and increasing levels of intramyocellular lipid content [26][27]. RA patients have an increased risk for developing insulin resistance, which is influenced by RF seropositivity, higher disease activity, prednisone use, and visceral and thigh intermuscular adiposity [24][28]. The increased accumulation of toxic lipid mediators and consequent lipotoxicity in skeletal muscles may also stimulate mitochondrial dysfunction, that, together with insulin resistance, contribute to cardiovascular disorders and sarcopenia [29]. Insulin resistance, cardiovascular disorders, and sarcopenia, associated with an increased intramuscular fat compared to healthy subjects, are frequently associated with RA [30].
Although the true etiology of this chronic inflammatory, rheumatic and immune-mediated disease remains unknown, several factors are known to contribute to the development and pathogenesis of RA, (Figure 1), namely inflammation exacerbation, oxidative stress and lipid peroxidation, as will be detailed in the following chapter.
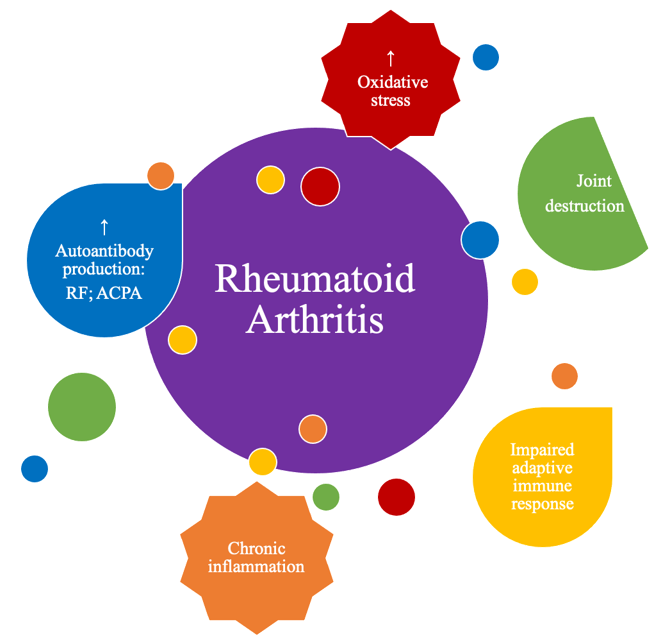
Figure 1. Main physio-pathological alterations that occur in rheumatoid arthritis (RA).
2. Lipidomic Studies in Rheumatoid Arthritis
From the evidence described in the previous chapter, it is accepted that, in fact, there are changes in the lipid metabolism in this pathology. The correct regulation of the lipid metabolism is vital for the homeostasis of the organism, thus avoiding pathological states. The dysregulation of lipid homeostasis affects several processes of inter and intracellular signalling and regulation of the immune response. Thus, the analysis of lipids and their variation is fundamental for a better understanding of the pathogenesis of several chronic diseases such as RA. Personalized medicine is the utmost tailored approach to disease management. Although the routine substances work for the general population, there are some patients to whom the drug effects are not the desired ones. To overcome this setback, biomarkers are of extreme importance to exclude the undesired effects of the drug treatment. These biomarkers may be autoantibodies, genes, or even biomolecules such as lipids. It should be noted that RA treatment involves corticosteroids, which are used as a powerful anti-inflammatory drug, but their effect in the lipid profile is unknown. Thus, clinical lipidomics is the utmost approach to evaluate the variation of lipids at a molecular level in several chronic diseases, including AID [31]. The aim of clinical lipidomics is to unveil the variation of the lipid profile in disease, to understand lipid metabolism in health and disease and identify new putative diagnostic biomarkers and therapeutic targets [32]. Clinical lipidomics is a reliable source of information regarding lipidic alterations and, hopefully, it is a promising approach to disease diagnostics and therapy monitorization in the near future.
Studies on the variation of RA lipid profile at a molecular level using lipidomics were gathered in this review. English language publications were identified through a computerized search (using PubMed database) until 2020 using the following keywords: lipidomic(s), lipid profile, fatty acid(s), phospholipid(s), sphingolipid(s), ceramide(s), and oxidized, combined with rheumatoid arthritis. Studies that did not report the use of mass spectrometry (MS) techniques were not taken into consideration.
2.1 Lipidomic Analysis of Serum/Plasma in Rheumatoid Arthritis Patients
Serum/plasma is the most important body fluid to portray metabolic changes. The analysis of serum/plasma sheds light on alterations induced by a pathological environment. The majority of the lipidomic studies on RA has been performed in human serum/plasma samples using mainly gas chromatography-mass spectrometry (GC-MS) for FA profiling, comparing RA patients with healthy controls, also including studies that evaluated the effect of FA diet, but not the effect of disease state. Few studies used liquid chromatography-mass spectrometry (LC-MS) for PL profiling, free fatty acids (FFA) and oxidized FA, also comparing data from plasma/serum from RA patients and controls.
2.1.1 Fatty Acid Profiling Analysis
The studies that reported the changes in FA profile in RA patients, compared with non-disease subjects, reported similar trends in some FA while opposite trends were reported in other studies. This can also be due to different experimental approaches since some studies investigate the variation of FA in plasma [33][34][35][36], while others reported in phosphatidylcholine (PC) [37] or in total PL [38][39][40][41][42]. The majority of the studies reported a decrease of ω-3 FA, namely a decrease of 18:3 ω-3, 20:5 ω-3 or/and 22:6 ω-3. A decrease of palmitic acid (16:0) was seen in some studies, but was increased in others, while the saturated FA 18:0 was reported to decrease or not change in different studies. The ω-6 FA, such as the arachidonic acid (20:4), showed also contradictory variations. Other FA were also affected by disease, as will be described.
Bruderlein and colleagues investigated the FA profile of serum PL of RA patients [38]. They determined significant decreases of FA 16:0; 18:2 ω-6 and 18:3 ω-3, and significant increases of FA 18:1 ω-9; 20:3 ω-6; 20:3 ω-9 and (20:3 + 20:4) ω-6/18:2 ω-6 ratio [38]. It was suggested that the ω-6 metabolic pathway is altered in RA patients, and it was induced by the persistent inflammation of this disease. The sum of ω-6 FA was constant, thus the disease seemed to stimulate an alteration in the content of different ω-6 FA towards higher unsaturated species, namely 20:3 and 20:4 [38]. When in a cell membrane, 20:3 ω-6 and 20:4 ω-6, have a much higher distortion, conformation wise, than that of 18:2 ω-6, due to their higher degree of unsaturation. Changes in the abundance of these FA may affect membrane properties [34]. Jacobsson and co-workers analyzed the FA composition of serum PC (the major PL class in plasma/serum lipid profile) in RA patients of short and long disease duration [37]. It was verified a significant decrease of FA 16:1; 18:0; 18:2 ω-6; 18:3 ω-3; 20:5 ω-3; 22:5 ω-3; 18:2/20:4 ratio and Σ ω-6 and a significant increase of FA 14:0; 16:0; 18:1 and Σ saturated FA. These changes became more evident with the increase of disease duration [37]. The disturbances in FA of the ω-6 series are in agreement with the previous findings of Bruderlein [38]. The reduced 18:2/20:4 ratio in RA patients was suggested to be related to the degree of inflammation, due to an increase in the FA desaturation/elongation process, which could be the result of higher insulin levels (stimulates desaturation) that have been found in RA patients, or due to an increased synthesis of eicosanoids [44][45]. Suryaprabha and colleagues studied plasma concentrations of essential FA of PL and determined a marked decrease of FA 18:0; 18:3 ω-6; 18:3 ω-3; 20:3 ω-6; 20:5 ω-3 and 22:6 ω-3 [39]. Interleukin-2 dependent T-cell proliferation and tumour necrosis factor and interleukin-2 production can be blocked by PUFA, thus, the reduction of essential FA and their metabolites may contribute to increase the chronic inflammation once T-cell proliferation, tumour necrosis factor and interleukin-2 production may not be inhibited [46]. The low levels of EPA and DHA also contribute to the inflammatory state [39].
Non-esterified FA/FFA profile of serum in RA was also evaluated by using LC-MS lipidomic approaches and revealed a significant reduction of FA 16:0; 16:1 ω-7; 18:1 ω-9 and 20:4 ω-6 when compared with healthy controls [33]. The FA 20:5 ω-3 and 22:6 ω-3 were reported to have significantly lower concentrations in RA patients as well [33]. These results were associated with SE presence, which suggests that non-esterified FA alterations could be on the onset of RA development.
2.1.2. FA Analysis after a Fasting Period
Changes in FA profile in RA with different diets or in a fasting/non-fasting regimen was also evaluated since fasting has been shown to have significant beneficial effects in clinical manifestations in RA [47]. Hafström and co-authors evaluated the serum levels of FA of membrane PL of RA patients after a fasting period [40] and observed markedly reduced levels of FA 20:3 ω-6 and markedly elevated levels of FA 20:4 ω-6 and 20:5 ω-3 comparing with RA patients before fasting and healthy control’s levels [40]. Haugen and colleagues investigated the possible impact on disease activity of fasting and one-year vegetarian diet in patients with RA [41]. The results showed that, after a vegan diet period, several FA of plasma PL were significantly decreased, such as FA 14:0; 16:0; 16:1; 18:0; 18:1; 18:3 ω-3; 20:1; 20:3 ω-9; 20:3 ω-6; 20:4 ω-6; 20:5 ω-3; 22:0; 22:1 ω-11; 22:4 ω-6; 22:5 ω-6; 22:5 ω-3; 22:6 ω-3; Σ ω-6; Σ ω-3 and Σ saturated FA [41]. A few years forward, Fraser and co-workers evaluated free FA changes in plasma of RA subjects after fasting and assessed the effects upon T lymphocyte proliferation [34]. Long chain ω-3 and ω-6 FA are linked with the inhibition of T lymphocyte function [48][49][50]. However, it is unclear if alterations on total concentration of circulating FFA would influence the immune response. Firstly, Fraser determined marked increases of FFA 14:0; 16:0; 16:1 ω-7; 18:0; 18:1 ω-9; 18:2 ω-6; 18:3 ω-3 and total FFA after a fasting period [34]. Secondly, it was shown that the proliferative response of T lymphocytes was higher with the increase of FA levels. Lastly, in vitro tests revealed that the ratio of unsaturated/saturated FFA had a significant effect upon lymphocyte proliferation. Lymphocyte proliferative responses, after mitogenic stimulation in the presence of 1) only unsaturated FA or 2) only saturated FA, were significantly lower than when stimulated in the presence of a mixture of unsaturated/saturated FA [34]. The practical implications of this finding to in vivo situations remains uncertain.
2.1.3. FA Analysis after Supplementation Intake
The effect of dietary fatty acid was also evaluated in a few studies, and dissimilar variations were found, which may be due to different supplements, methods, and samples. Kremer and co-workers examined the effect of manipulation of dietary FA comparing plasma of RA patients with and without EPA supplementation [35]. As expected, patients taking EPA supplementation had significantly higher levels of plasma EPA than the patients not taking the supplementation [35]. Remans and co-authors had similar results as obtained by Kremer’s team. Remans compared the FA profile of PL of active RA patients receiving nutrient supplementation containing PUFA, including EPA, and micronutrients, with a group of active RA patients receiving placebo [42]. All patients receiving nutrient supplementation had significant increases of total ω-3 PUFA (20:5; 22:5; 22:6) and decrease of AA [42]. Although there were significant changes in plasma FA, the nutritional supplementation did not improve signs and symptoms of RA patients. On the other side, Jäntti and colleagues also evaluated the influence of supplementation with evening primrose oil (rich in FA 18:3 ω-6) and determined significantly lower concentrations of serum FA 18:1 and EPA; and markedly increased levels of serum FA 18:2; 18:3; 20:3 and 20:4 [36]. The intake of evening primrose oil had contradictory effects, increasing both anti-inflammatory FA 18:3 and pro-inflammatory AA. The increase of FA 18:3 may be regarded as favorable to help reduce the inflammatory status. However, the increase of AA may be considered as harmful since it is the precursor of pro-inflammatory prostaglandins and leukotrienes [36]. Proudman [51] analyzed the results of an investigation conducted previously by other authors, and therefore did not mention original results to be discussed in this review. PUFA supplementation should have, in theory, beneficial results and improve clinical symptoms because EPA and DHA compete with AA for incorporation in cellular membranes, which leads to a reduction in the synthesis of prostaglandins and leukotrienes, thus reducing the inflammatory state [52]. However, investigators struggle to find the correct dosage of PUFA supplementation proven by different studies (with unequal doses) having different results. Having said that, more studies are needed to ascertain the quantity of PUFA supplementation should be administered to RA patients in order to maximize the results.
2.1.4. Phospholipidomic Profiling Analysis by LC-MS
To our knowledge, there is only one study that reported untargeted lipidomic analysis of plasma PL of RA subjects. Łuczaj et al. [53] determined alterations on the PL profile that reflected a decrease on PC(40:2); PC(40:3) and PC(42:3), and an increase on lyso-PC (LPC) species LPC(16:1); LPC(24:3); PC(34:3); PC(36:3); PC(38:2); PC(38:3); PC(38:4); lyso-phosphatidylethanolamine (LPE) species LPE(16:0); LPE(18:0); phosphatidylethanolamine (PE) species PE(30:1); phosphatidylinositol (PI) species PI(36:1); PI(36:2); PI(36:3); PI(36:4); PI(38:3); PI(38:4) and sphingomyelin (SM) species SM(d34:2); SM(d38:1); SM(d40:1) and SM(d40:2) [53]. The results suggest a significantly altered PE and PC metabolism with enhanced PL hydrolysis by phospholipase A2. In addition, the increase of LPC in RA has already been associated with the induction of cyclooxygenase 2 hence contributing to systemic inflammation [54][55]. PC and PE can also be modified by ROS in oxidative stress conditions [56]. Although the main targets of oxidation are the unsaturated FA chains and PC is more abundant than PE, PE is more reactive over PC since it has a free amino group in the polar head, being a preferable target to suffer modifications induced by oxidative stress conditions and ROS which are enhanced in inflammation. Sphingolipids have pleotropic pro-inflammatory effects. Therefore, the study of its metabolism is very important. Regarding sphingolipids metabolism, other findings worth mentioning are the detection of increased levels of total monohexosylceramides (HexCer, as a sum of HexCer16:0; HexCer16:1; HexCer18:0; HexCer20:0; HexCer22:0; HexCer23:0; HexCer24:0 and HexCer24:1), total ceramides (Cer, as a sum of Cer16:0; Cer18:0; Cer20:0; Cer22:0; Cer23:0; Cer24:0; Cer24:1; Cer25:0 and Cer26:0;) and sphingosine (d18:1) in serum of established RA patients [57].
2.1.5. Identification of Lipid Peroxidation Products by LC-MS
Lipid peroxidation products of AA and linoleic acid in RA patients were evaluated through LC-MS techniques by Charles-Schoeman and co-workers [58]. It was analyzed the presence of hydroxyeicosatetraenoic acids (HETE) and hydroxyoctadecadienoic acids (HODE) in plasma lipoproteins HDL and LDL. The results showed markedly increase levels of oxidation products 5-HETE; 15-HETE; 9-HODE and 13-HODE in both HDL and LDL fractions of RA patients [58]. HETE and HODE contribute to LDL oxidation which, together with their accumulation in HDL particles, may inhibit HDL beneficial function increasing the risk of developing atherosclerotic events [59][60]. Higher concentrations of these lipid peroxidation products in HDL are associated with the decreased antioxidant capacity of such particles. Thus, the raised levels of systemic inflammation, also determined in this investigation, positively correlates with the increased levels of free oxidized FA in HDL and LDL, reassuring the fact that RA is an AID with an exacerbated oxidative environment [58].
3. Concluding remarks
Overall, the serum/plasma lipidomic analysis of the studies reported above suggest that RA is characterized by an altered lipid profile. Plasma FA alterations were observed and revealed as significant lower levels of FA 20:5 ω-3 and 22:6 ω-3, although some contradictory variations were reported, as for other FA. The plasma PL profile was found to be changed with manifestations on PE and PC metabolism with enhanced PL hydrolysis, in a solely study that used lipidomic approaches. Some attempts were made regarding the effects of PUFA supplementation to compensate the lower level of ω-3 PUFA in RA, but more studies are needed to understand exactly which dose should be administrated and if, besides FA alterations, there are, in fact, changes concerning disease activity. Nonetheless, lipid metabolism is undeniably affected in RA. Considering the important role of lipids in inflammation, more studies are needed, particularly using modern lipidomics, which could contribute to unveil the pathophysiology of this disease, find new biomarkers, and to develop better therapeutic approaches.
References
- Alamanos, Y.; Voulgari, P.V.; Drosos, A.A. Incidence and prevalence of rheumatoid arthritis, based on the 1987 American College of Rheumatology criteria: A systemic review. Arthritis Rheum. 2006, 36, 182–188.
- Sandoughi, M.; Kaykhaei, M.A.; Shahrakipoor, M.; Darvishzadeh, R.; Nikbakht, M.; Shahbakhsh, S.; Zakeri, Z. Clinical manifestations and disease activity score of rheumatoid arthritis in southeast of Iran. Res. 2017, 2, 61–64.
- Jung, N.; Bueb, J.L.; Tolle, F.; Bréchard, S. Regulation of neutrophil pro-inflammatory functions sheds new light on the pathogenesis of rheumatoid arthritis. Pharmacol. 2019, 165, 170–180.
- Aletaha, D.; Blüml, S. Therapeutic implications of autoantibodies in rheumatoid arthritis. RMD Open 2016, 2, e000009.
- Aggarwal, R.; Liao, K.; Nair, R.; Ringold, S.; Costenbader, K.H. Anti-citrullinated peptide antibody assays and their role in the diagnosis of rheumatoid arthritis. Arthritis Rheum. 2009, 61, 1472–1483.
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O.; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581.
- Bartok, B.; Firestein, G.S. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Rev. 2010, 233, 233–255.
- Marston, B.; Palanichamy, A.; Anolik, J.H. B cells in the pathogenesis and treatment of rheumatoid arthritis. Opin. Rheumatol. 2010, 22, 307–315.
- Su, Z.; Yang, R.; Zhang, W.; Xu, L.; Zhong, Y.; Yin, Y.; Cen, J.; DeWitt, J.P.; Wei, Q. The synergistic interaction between the calcineurin B subunit and IFN-γ enhances macrophage antitumor activity. Cell Death Dis. 2015, 6, e1740.
- Tolboom, T.C.; Pieterman, E.; Laan, W.H. Van Der; Toes, R.E.; Huidekoper, A.L.; Nelissen, R.G.; Breedveld, F.C.; Huizinga, T.W. Invasive properties of fibroblast-like synoviocytes: Correlation with growth characteristics and expression of MMP-1, MMP-3, and MMP-10. Rheum. Dis. 2002, 61, 975–980.
- Sabeh, F.; Fox, D.; Weiss, S.J. Membrane-type I matrix metalloproteinase-dependent regulation of rheumatoid arthritis synoviocyte function. Immunol. 2010, 184, 6396–6406.
- Smiljanovic, B.; Radzikowska, A.; Kuca-Warnawin, E.; Kurowska, W.; Grün, J.R.; Stuhlmüller, B.; Bonin, M.; Schulte-Wrede, U.; Sörensen, T.; Kyogoku, C.; et al. Monocyte alterations in rheumatoid arthritis are dominated by preterm release from bone marrow and prominent triggering in the joint. Rheum. Dis. 2018, 77, 300–308.
- Cascão, R.; Moura, R.A.; Perpétuo, I.; Canhão, H.; Vieira-Sousa, E.; Mourão, A.F.; Rodrigues, A.M.; Polido-Pereira, J.; Queiroz, M.V.; Rosário, H.S.; et al. Identification of a cytokine network sustaining neutrophil and Th17 activation in untreated early rheumatoid arthritis. Arthritis Res. Ther. 2010, 12, R196.
- Wright, H.L.; Moots, R.J.; Bucknall, R.C.; Edwards, S.W. Neutrophil function in inflammation and inflammatory diseases. Rheumatology 2010, 49, 1618–1631.
- Larbre, J.P.; Moore, A.R.; Silva, J.A. Da; Iwamura, H.; Ioannou, Y.; Willoughby, D.A. Direct degradation of articular cartilage by rheumatoid synovial fluid: Contribution of proteolytic enzymes. Rheumatol. 1994, 21, 1796–1801.
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Transl. Med. 2013, 5, 178ra40.
- Bala, A.; Mondal, C.; Haldar, P.K.; Khandelwal, B. Oxidative stress in inflammatory cells of patient with rheumatoid arthritis: Clinical efficacy of dietary antioxidants. Inflammopharmacology 2017, 25, 595–607.
- Karami, J.; Aslani, S.; Jamshidi, A.; Garshasbi, M.; Mahmoudi, M. Genetic implications in the pathogenesis of rheumatoid arthritis; an updated review. Gene 2019, 702, 8–16.
- Takeno, M.; Kitagawa, S.; Yamanaka, J.; Teramoto, M.; Tomita, H.; Shirai, N.; Itoh, S.; Hida, S.; Hayakawa, K.; Onozaki, K.; et al. 5-Hydroxy-2-methylpyridine isolated from cigarette smoke condensate aggravates collagen-induced arthritis in mice. Pharm. Bull. 2018, 41, 877–884.
- Croia, C.; Bursi, R.; Sutera, D.; Petrelli, F.; Alunno, A.; Puxeddu, I. One year in review 2019: Pathogenesis of rheumatoid arthritis. Exp. Rheumatol. 2019, 37, 347–357.
- Knevel, R.; De Rooy, D.P.; Saxne, T.; Lindqvist, E.; Leijsma, M.K.; Daha, N.A.; Koeleman, B.P.; Tsonaka, R.; Houwing-Duistermaat, J.J.; Schonkeren, J.J. A genetic variant in osteoprotegerin is associated with progression of joint destruction in rheumatoid arthritis. Arthritis Res. Ther. 2014, 16, R108.
- de Rooy, D.P.; Tsonaka, R.; Andersson, M.L.; Forslind, K.; Zhernakova, A.; Frank-Bertoncelj, M.; de Kovel, C.G.; Koeleman, B.P.; van der Heijde, D.M.; Huizinga, T.W. Genetic factors for the severity of ACPA-negative rheumatoid arthritis in 2 cohorts of early disease: A genome-wide study. Rheumatol. 2015, 42, 1383–1391.
- Van Vollenhoven, R.F. Progress in RA genetics, pathology and therapy. Rev. Rheumatol. 2013, 9, 70–72.
- Hanaoka, B.Y.; Ithurburn, M.P.; Rigsbee, C.A.; Bridges, S.L.; Moellering, D.R.; Gower, B.; Bamman, M. Chronic inflammation in RA: Mediator of skeletal muscle pathology and physical impairment. Arthritis Care Res. 2020, 71, 173–177.
- Huffman, K.M.; Jessee, R.; Andonian, B.; Davis, B.N.; Narowski, R.; Huebner, J.L.; Kraus, V.B.; McCracken, J.; Gilmore, B.F.; Tune, K.N.; et al. Molecular alterations in skeletal muscle in rheumatoid arthritis are related to disease activity, physical inactivity, and disability. Arthritis Res. Ther. 2017, 19, 12.
- VanderVeen, B.N.; Fix, D.K.; Carson, J.A. Disrupted skeletal muscle mitochondrial dynamics, mitophagy, and biogenesis during cancer cachexia: A role for inflammation. Med. Cell. Longev. 2017, 2017, 3292087.
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. Engl. J. Med. 2004, 350, 664–671.
- Giles, J.T.; Danielides, S.; Szklo, M.; Post, W.S.; Blumenthal, R.S.; Petri, M.; Schreiner, P.J.; Budoff, M.; Detrano, R.; Bathon, J.M. Insulin resistance in rheumatoid arthritis: Disease-related indicators and associations with the presence and progression of subclinical atherosclerosis. Arthritis Rheumatol. 2015, 67, 626–636.
- Lanchais, K.; Capel, F.; Tournadre, A. Could omega 3 fatty acids preserve muscle health in rheumatoid arthritis? Nutrients 2020, 12, 223.
- Baker, J.F.; Mostoufi-Moab, S.; Long, J.; Zemel, B.; Ibrahim, S.; Taratuta, E.; Leonard, M.B. Intramuscular fat accumulation and associations with body composition, strength, and physical functioning in patients with rheumatoid arthritis. Arthritis Care Res. 2018, 70, 1727–1734.
- Zhao, Y.Y.; Cheng, X. long; Lin, R.C. Lipidomics Applications for Discovering Biomarkers of Diseases in Clinical Chemistry; Int. Rev. Cell Mol. Biol. 2014; 313, 1-26.
- Lv, J.; Zhang, L.; Yan, F.; Wang, X. Clinical lipidomics: A new way to diagnose human diseases. Clin. Transl. Med. 2018, 7, 10–12.
- Rodríguez-Carrio, J.; Alperi-López, M.; López, P.; Ballina-García, F.J.; Suárez, A. Non-esterified fatty acids profiling in rheumatoid arthritis: Associations with clinical features and Th1 response. PLoS ONE 2016, 11, 1–17.
- Fraser, D.A.; Thoen, J.; Rustan, A.C.; Førre, Ø.; Kjeldsen-Kragh, J. Changes in plasma free fatty acid concentrations in rheumatoid arthritis patients during fasting and their effects upon T-lymphocyte proliferation. Rheumatology 1999, 38, 948–952.
- Kremer, J.M.; Michalek, A.V.; Lininger, L.; Huyck, C.; Bigauoette, J.; Timchalk, M.A.; Rynes, R.I.; Zieminski, J.; Bartholomew, L.E. Effects of manipulation of dietary fatty acids on clinical manifestations of rheumatoid arthritis. Lancet 1985, 1, 184–187.
- Jäntti, J.; Nikkari, T.; Solakivi, T.; Vapaatalo, H.; Isomaki, H. Evening primrose oil in rheumatoid arthritis: Changes in serum lipids and fatty acids. Ann. Rheum. Dis. 1989, 48, 124–127.
- Jacobsson, L.; Lindgarde, F.; Manthorpe, R.; Akesson, B. Correlation of fatty acid composition of adipose tissue lipids and serum phosphatidylcholine and serum concentrations of micronutrients with disease duration in rheumatoid arthritis. Ann. Rheum. Dis. 1990, 49, 901–905.
- Bruderlein, H.; Daniel, R.; Boismenu, D.; Julien, N.; Couture, F. Fatty acid profiles of serum phospholipids in patients suffering rheumatoid arthritis. Prog. Lipid Res. 1981, 20, 625–631.
- Suryaprabha, P.; Das, U.N.; Ramesh, G.; Kumar, K.V.; Kumar, G.S. Reactive oxygen species, lipid peroxides and essential fatty acids in patients with rheumatoid arthritis and systemic lupus erythematosus. Prostaglandins Leukot. Essent. Fat. Acids 1991, 43, 251–255.
- Hafström, I.; Ringertz, B.; Gyllenhammar, H.; Palmblad, J.; Harms‐Ringdahl, M. Effects of fasting on disease activity, neutrophil function, fatty acid composition, and leukotriene biosynthesis in patients with rheumatoid arthritis. Arthritis Rheum. 1988, 31, 585–592.
- Haugen, M.A.; Kjeldsen-Kragh, J.; Bjervea, K.S.; Høstmark, A.T.; Førre, Ø. Changes in plasma phospholipid fatty acids and their relationship to disease activity in rheumatoid arthritis patients treated with a vegetarian diet. Br. J. Nutr. 1994, 72, 555–566.
- Remans, P.H.J.; Sont, J.K.; Wagenaar, L.W.; Wouters-Wesseling, W.; Zuijderduin, W.M.; Jongma, A.; Breedveld, F.C.; van Laar, J.M. Nutrient supplementation with polyunsaturated fatty acids and micronutrients in rheumatoid arthritis: Clinical and biochemical effects. Eur. J. Clin. Nutr. 2004, 58, 839–845.
- Ferber, E.; De Pasquale, G.G.; Resch, K. Phospholipid metabolism of stimulated lymphocytes. Composition of phospholipd fatty acids. Biochim. Biophys. Acta. 1975, 398, 364–376.
- Svensson, K.; Lundqvist, G.; Wide, L.; Hallgren, R. Impaired glucose handling in active rheumatoid arthritis: Relationship to the secretion of insulin and counter-regulatory hormones. Metabolism 1987, 36, 944–948.
- Brenner, R. Nutritional and hormonal factors influencing desaturation of essential fatty acids. Prog. Lipid Res. 1982, 20, 41–48.
- Santoli, D.; Phillips, P.D.; Colt, T.L.; Zurier, R.B. Suppression of interleukin-2 dependent human T cell growth in vitro by Prostaglandin E and their precursor fatty acids. J. Clin. Invest. 1990, 85, 424–432.
- Athanassiou, P.; Athanassiou, L.; Kostoglou-Athanassiou, I. Nutritional pearls: Diet and rheumatoid arthritis. Mediterr. J. Rheumatol. 2020, 31, 319–324.
- Rossetti, R.; Seiler, C.; DeLuca, P.; Laposata, M.; Zurier, R. Oral administration of unsaturated fatty acids: Effects on human peripheral blood T lymphochyte proliferation. J. Leukoc. Biol. 1997, 62, 438–443.
- Purasiri, P.; McKechnie, A.; Heys, S.; Eremin, O. Modulation in vitro of human natural cytotoxicity, lymphocyte proliferative response to mitogens and cytokine production by essential fatty acids. Immunology 1997, 92, 166–172.
- Søyland, E.; Nenseter, M.; Braathen, L.; Drevon, C. Very long chain n-3 and n-6 polyunsaturated fatty acids inhibit proliferation of human T-lymphocytes in vitro. Eur. J. Clin. Investig. 1993, 23, 112–121.
- Proudman, S.M.; Cleland, L.G.; Metcalf, R.G.; Sullivan, T.R.; Spargo, L.D.; James, M.J. Plasma n-3 fatty acids and clinical outcomes in recent-onset rheumatoid arthritis. Br. J. Nutr. 2015, 114, 885–890.
- Kremer, J.M. n-3 fatty acid supplements in rheumatoid arthritis. Am. J. Clin. Nutr. 2000, 71, 349S–351S.
- Łuczaj, W.; Moniuszko-Malinowska, A.; Domingues, P.; Domingues, M.R.; Gindzienska-Sieskiewicz, E.; Skrzydlewska, E. Plasma lipidomic profile signature of rheumatoid arthritis versus Lyme arthritis patients. Arch. Biochem. Biophys. 2018, 654, 105–114.
- Fuchs, B.; Schiller, J.; Wagner, U.; Häntzschel, H.; Arnold, K. The phosphatidylcholine/ lysophosphatidylcholine ratio in human plasma is an indicator of the severity of rheumatoid arthritis: Investigations by 31 P NMR and MALDI-TOF MS. Clin. Biochem. 2005, 38, 925–933.
- Rikitake, Y.; Hirata, K.; Kawashima, S.; Takeuchi, S.; Shimokawa, Y.; Kojima, Y.; Inoue, N.; Yokoyama, M. Signaling mechanism underlying COX-2 induction by lysophosphatidylcholine. Biochem. Biophys. Res. Commun. 2001, 281, 1291–1297.
- Domingues, M.R.M.; Reis, A.; Domingues, P. Mass spectrometry analysis of oxidized phospholipids. Chem. Phys. Lipids 2008, 156, 1–12.
- Miltenberger-Miltenyi, G.; Cruz-Machado, A.R.; Saville, J.; Conceição, V.A.; Calado, Â.; Lopes, I.; Fuller, M.; Fonseca, J.E. Increased monohexosylceramide levels in the serum of established rheumatoid arthritis patients. Rheumatology 2020, 59, 2085–2089.
- Charles-Schoeman, C.; Meriwether, D.; Lee, Y.Y.; Shahbazian, A.; Reddy, S.T. High levels of oxidized fatty acids in HDL are associated with impaired HDL function in patients with active rheumatoid arthritis. Clin. Rheumatol. 2018, 37, 615–622.
- Morgantini, C.; Natali, A.; Boldrini, B.; Imaizumi, S.; Navab, M.; Fogelman, A.M.; Ferrannini, E.; Reddy, S.T. Anti-inflammatory and antioxidant properties of HDLs are impaired in type 2 diabetes. Diabetes 2011, 60, 2617–2623.
- Imaizumi, S.; Grijalva, V.; Navab, M.; Lenten, B.J. Van; Wagner, A.C.; Anantharamaiah, G.M.; Fogelman, A.M.; Reddy, S.T. L-4F differentially alters plasma levels of oxidized fatty acids resulting in more anti-inflammatory HDL in mice. Drug Metab. Lett. 2010, 4, 139–148.


