1000/1000
Hot
Most Recent
 +1 point
+1 point
Oxidative stress is defined as an imbalance between the cellular levels of antioxidants and that of pro-oxidants, including ROS and reactive nitrogen species, which causes cellular damage and, in most cases, cell death. Non-alcoholic fatty liver disease (NAFLD) is often the hepatic expression of metabolic syndrome and its comorbidities that comprise, among others, obesity and insulin-resistance. At the molecular level, several models have been proposed for the pathogenesis of NAFLD. Most importantly, oxidative stress and mitochondrial
damage have been reported to be causative in NAFLD initiation and progression.
Non-alcoholic fatty liver disease (NAFLD) frequently co-exists with metabolic syndrome and thus often is defined as the liver expression of dyslipidemia, insulin resistance, and obesity. However, it can also occur in patients with low body mass index (<25 kg/m2), a condition known as lean NAFLD [1][2][3]. NAFLD is characterized by fat accumulation in hepatocytes and comprises a spectrum of disease. This spectrum ranges from simple steatosis (non-alcoholic fatty liver or NAFL) to non-alcoholic steatohepatitis (NASH) with inflammation, hepatocyte ballooning, and various levels of fibrosis, associated with a significant risk for progression to cirrhosis and liver cancer (hepatocellular carcinoma (HCC)). In fact, only 20% of NAFLD patients develop NASH, and 4% of patients with NAFL and 20% of patients with NASH develop cirrhosis [4]. Over time, up to 5% of patients with NASH will experience spontaneous regression. NASH is strongly associated with the degree of hepatic fibrosis, which is predictive of the overall mortality in patients with NAFLD[4].
NAFLD prevalence is increasing among children [5][6] and has become the most common chronic liver disease in childhood and adolescence, as it is among adults. NAFLD affects approximately 25% of the global population [7], but the prevalence varies considerably by region. It is highest in the Middle East (32%) and South America (30%) and lowest in Africa (13%). In North America and Europe, the prevalence is 24%, and it is 27% in Asia[7]. As the overweight and obesity pandemic drives the development of metabolic conditions promoting NAFLD, the health and economic burdens continuously increase, also fueled by the boom in childhood obesity and an aging population[8][9].
NAFLD is defined as >55 mg triglycerides (TGs) per gram of liver or the presence of more than 5% steatotic hepatocytes without histological damage or inflammation. Most commonly, the percentage of hepatocytes presenting lipid accumulation is divided into three ranges: 5% to 33%, 34% to 66%, and >66%. Accordingly, the severity of steatosis is considered to be mild, moderate, or severe, respectively[10]. NAFL can progress to NASH that is characterized, in addition to steatosis, by ballooning degeneration of hepatocytes and lobular inflammation. The presence of perisinusoidal fibrosis is generally not considered a prerequisite for NASH diagnosis[11]. A recent study has found that some patients with NAFLD but without NASH may develop progressive fibrosis and have an increased mortality risk regardless of their baseline NASH status[12]. Furthermore, fibrosis progression is faster in lean patients with NAFLD, who can have a higher risk of developing cirrhosis or HCC compared with NAFLD patients who are overweight[13]. This pattern suggests that once the disease has progressed to NASH, obesity may not be the key factor in fibrosis progression [14], but this association remains debated and needs further investigation.
Oxidative stress is defined as an imbalance between the cellular levels of antioxidants and that of pro-oxidants, including ROS and reactive nitrogen species (Figure 1), which causes cellular damage and, in most cases, cell death. In healthy physiological conditions, cells maintain a basal level of ROS to promote the balanced redox signaling required for various processes such as cell metabolism, cell differentiation and survival, immune defense, and modulation of transcription factor activity and epigenetic state[15]. Upon oxidative stress, the antioxidant enzyme superoxide dismutase (SOD) generates hydrogen peroxide (H2O2) from the superoxide radical (O2.−), which is then processed to oxygen (O2) and water (H2O) through glutathione peroxidase (Gpx) or catalase enzyme activities (Figure 2, figure 3 and figure 5)[16]. Within cells, ROS are mainly produced in the mitochondria, the peroxisomes, and the ER, but cytoplasmic production of ROS also occurs. High levels of ROS alter these organelles, further enhancing oxidative stress and creating a vicious circle.
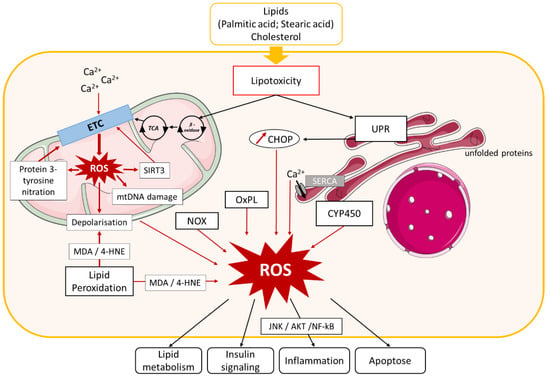
Figure 1. Role of oxidative stress in NAFLD. Lipid and cholesterol accumulation in hepatocytes causes lipotoxicity, resulting in activation of different pathways involved in oxidative stress. Lipid entry and alteration of mitochondrial Ca2+ homeostasis cause electron transport chain (ETC) dysfunction that leads to mitochondrial reactive oxygen species (ROS) production, which in turn induces Sirtuin (SIRT)3 expression, protein 3-tyrosine nitration, mitochondrial DNA damage, and membrane depolarization. SIRT3 and protein nitration also cause ETC dysfunction and exacerbate mitochondrial ROS production. Mitochondrial biogenesis dysfunction additionally induces ROS production. Lipotoxicity activates endoplasmic reticulum (ER) stress, leading to ROS production by (i) unfolded protein response (UPR) activation, which stimulates C/EBP homologous protein (CHOP) expression and (ii) Ca2+ homeostasis dysfunction. ROS generation in cytoplasm is induced by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) enzyme activation, oxidized phospholipids (OxPL), several cytochrome p450 enzymes (CYP450), and lipid peroxidation. ROS accumulation in hepatocytes leads to impairment of several pathways involved in NAFLD development, such as lipid metabolism, insulin signaling, inflammation, and apoptosis. Other abbreviations: mtDNA, mitochondrial DNA; TCA, tricarboxylic acid cycle; MDA, malondialdehyde; 4-HNE, 4-hydroxynonenal; SERCA, ER calcium pump sarco/endoplasmatic reticulum Ca2+-ATPase; JNK, c-Jun N-terminal kinase; NF-κB, nuclear factor-kappa B.
Oxidative stress promotes activation of enzymatic or non-enzymatic-mediated antioxidant mechanisms that counteract ROS production. Both clinical and experimental studies show that these antioxidant pathways are modulated during NAFLD progression. In fact, activity of the antioxidant enzymes SOD and Gpx increases in patients with NAFLD (Figure 2, Figure 3 , figure 4 and Figure 5)[17]. In vitro, hepatic stellate cells deficient for the glutathione peroxidase 7 (Gpx7) isoform present increased expression of profibrotic and pro-inflammatory genes in response to FFA exposure. Consistent with these results, overexpression of GPX7 in these cells decreases ROS generation and expression of profibrotic and pro-inflammatory genes. In vivo, Gpx7 deficiency exacerbates choline-deficient, L-amino–defined, high-fat diet-induced NASH fibrosis[18]. The expression of glutaminase 1 (GLS1) is increased in both NASH preclinical mouse models and clinical NASH liver biopsies. GLS1 promotes glutamine fueling of anaplerotic mitochondrial metabolism resulting in increased ROS production. In methionine choline-deficient (MCD) diet-fed mice, GLS1 inhibition decreases hepatic TG accumulation by restoring VLDL TG export and diminishes oxidative stress by lowering ROS production. GLS1 deficiency in this model is also associated with decreased lipid peroxidation [19]. Paraoxonase-1 is a liver antioxidant enzyme that hydrolyses peroxide and lactones associated with lipoproteins. In a cohort of 81 patients with NAFLD, the serum Paraoxonase-1 concentration was decreased, which could reflect higher oxidative stress in these patients [20].
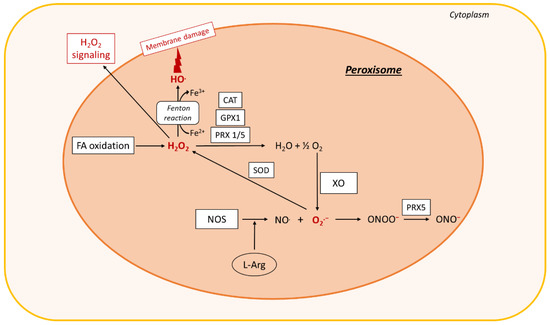
Figure 2. ROS production in peroxisomes. In peroxisomes, fatty acid (FA) oxidation leads to the formation of H2O2, which is decomposed by the antioxidant enzymes GPX1, CAT, or peroxiredoxin (PRX) 1 or 5, leading to the formation of O2 and H2O. O2 can react with XO to produce O2.−. Nitric oxide synthase (NOS) interaction with L-arginine (L-Arg) leads to the formation of NO., which interacts with O2.− to form ONOO−. ONOO− is converted by Prx5 to a peroxynitrite radical ONO.. SOD2 dismutates O2.− to H2O2. The Fenton reaction also occurs in peroxisomes and leads to membrane damage. The H2O2 is then exported to the cytoplasm, where it acts as a signaling molecule that exacerbates oxidative stress. Other abbreviations: XO, xanthine oxidase; CAT, catalase; GPX1, glutathione peroxidase 1; SOD, superoxide dismutase; Fe2+, ferrous iron; O2, oxygen; H2O, water; NO., nitric oxide; ONOO−, peroxynitrite; HO., hydroxyl radical; ONO., peroxynitrite radical; O2.−, superoxide; H2O2, hydrogen peroxide.
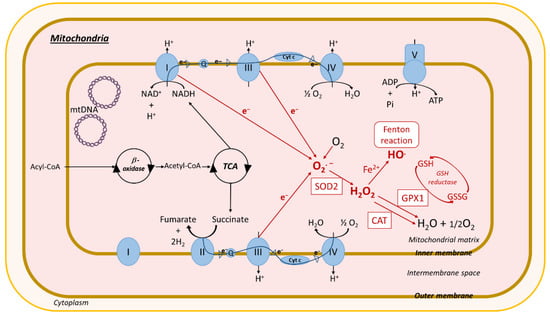
Figure 3. ROS production in mitochondria. Acyl-CoA imported into the mitochondria is converted by β-oxidation into acetyl-CoA, which enters the tricarboxylic acid cycle (TCA), resulting in the production of nicotinamide adenine dinucleotide (NADH) and succinate. Mitochondria have two respiratory chains composed of different complexes: (i) complexes I (NADH-ubiquinone oxidoreductase), III (cytochrome b-c1), and IV (cytochrome c oxidase). Complex I reduces NADH to NAD+ and H+ after electron (e−) release, and (ii) complexes II (succinate-quinone oxidoreductase), III, and IV. Complex II catalyzes the dehydrogenation and oxidation of succinic acid into furamate and 2H2 after e−release. Released electrons pass through complexes and other molecules in the mitochondria inner membrane, ubiquinone (Q) and cytochrome c (cyt c). Complex V (F1F0-ATP synthase) uses the chemiosmotic proton gradient to power the synthesis of ATP from adenosine-diphosphate (APD) and Pi. In red: impairment of the electron transport chain results in the leakage of e− that react directly with oxygen to form the superoxide anion radical transformed in H2O2 through SOD2 activity. H2O2 can react with Fe2+ to form OH. (Fenton reaction). H2O2 is processed into H2O and O2 by the antioxidant enzymes GPX1 and catalase. Mitochondrial ROS production causes oxidative mtDNA, lipid, and protein damage. Other abbreviations: CAT, catalase; GPX1, glutathione peroxidase 1; SOD2, superoxide dismutase; Fe2+, ferrous iron; O2, oxygen; H2O, water; HO., hydroxyl radical; O2.−, superoxide; H2O2, hydrogen peroxide; ATP, adenosine-triphosphate; Pi, inorganic phosphate; NAD+, nicotinamide adenine dinucleotide; H+, hydrogen; mtDNA, mitochondrial DNA; GSH, glutathione; GSSG, oxidized glutathione; ROS, reactive oxygen species.
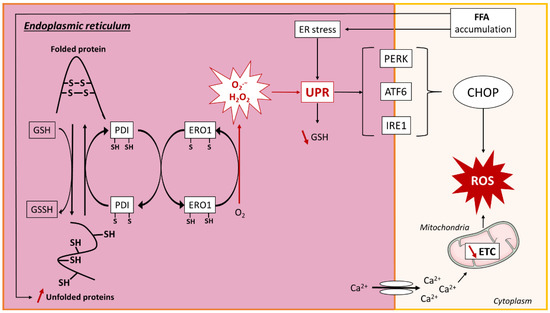
Figure 4. ROS production in endoplasmic reticulum. Lipid overload causes accumulation of unfolded proteins in the endoplasmic reticulum (ER) by the reduction of reduced glutathione (GSH) to oxidized glutathione (GSSH), resulting in the breakage of protein disulfide bonds (SH). The ER resident proteins, protein disulfide isomerase (PDI) and ER oxidoreductin 1 (ERO1), are involved in disulfide bond formation and superoxide (O2.−) and hydrogen peroxide (H2O2) release through electron transfer. ROS production and ER stress activate the unfolded protein response (UPR), which leads to a decrease in GSH. UPR is regulated by three transmembrane proteins: protein kinase RNA-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring signaling protein 1 (IRE1), which promote the transcription of CCAAT/enhancer-binding protein homologous protein (CHOP) that further induces ROS generation. ER stress is associated with reduced sarco/endoplasmatic reticulum Ca2+-ATPase (SERCA) activity, leading to calcium leakage and decreased mitochondrial electron transport chain (ETC) activity, resulting in ROS production. Other abbreviations: O2, oxygen; FFA, free fatty acid; ROS, reactive oxygen species; Ca2+, calcium.
The peroxisomal antioxidant enzyme catalase plays a key role in protecting cells from oxidative damage by reducing H2O2 concentration (Figure 2). In HFD-fed mice deficient for catalase, lipid accumulation and oxidative stress are exacerbated[21]. These mice develop an imbalanced redox status in peroxisomes because of increased H2O2 levels, which in turn induces ER stress in the liver. Catalase inhibition in HepG2 cells increases ROS production by peroxisomes, causing ER stress and FA accumulation[22]. In line with this observation, human liver cells with impaired peroxisome biogenesis show decreased ER stress, oxidative stress, and apoptosis [23].
Expression of genes encoding antioxidant enzymes and regulators of the glutathione pathway (glutamate-cysteine ligase catalytic subunit, glutamate-cysteine ligase modifier subunit, GPX2) is regulated by the transcription factor erythroid2-like 2 (Nrf2)[24]. Upon oxidative stress, Nrf2, which is anchored in the cytoplasm via binding to Keap1, dissociates from it and translocates into the nucleus, where it interacts with specific DNA sequences called antioxidant response elements in the promoter of its target antioxidant enzyme genes [25]. Nrf2 expression is increased in the first stage of NAFLD in preclinical models [26], and pharmacological activation of Nrf2 in mice fed a high-fat and high fructose diet decreases NASH parameters (insulin resistance, weight gain, TG, ALT) via the transcriptional regulation of genes involved in inflammation, apoptosis, fibrosis, ER stress, and oxidative stress [24]. Consistently, Nrf2 deficiency in mice fed an MCD or an HFD promotes progression of steatosis to NASH by increasing oxidative stress, inflammation, and hepatic FA accumulation [27][28]. In addition, Nrf2-deficient mice fed an HFD develop a more severe NASH phenotype than their WT counterparts [26]. In in vivo and in vitro models of NAFLD, the reduction of Nrf2 expression by microRNA is associated with a decreased expression of its target genes, heme oxygenase, Sod2, and NAD(P)H dehydrogenase quinone 1 and an increase in ROS production [29]. Nrf2 also directly affects lipid metabolism by activation of genes involved in FA oxidation (acyl-CoA Oxidase 2, carnitine palmitoyltransferase 1), TG export (apolipoprotein B), and the lipogenic transcription factor sterol regulatory element binding transcription factor 1 (Srebp-1)[24]. Nfr2 deficiency in HFD-fed mice diminishes phosphorylation of acetyl-CoA carboxylate (ACC), a rate-limiting enzyme of hepatic FA synthesis, and thus increases its activity [26]. Collectively, these data indicate that alterations in antioxidant pathways are associated with NAFLD, suggesting a role of oxidative stress in disease progression.
Mitochondria are intracellular sites of oxygen (O2) consumption. In case of energy imbalance, such as lipid accumulation, mitochondria ROS production increases considerably because of mitochondrial respiratory chain alteration and a reduction in electron capture through various mechanisms discussed below (Figure 3). Generation of mitochondrial ROS results in impairment of the mitochondrial membrane potential generated by proton pumps and activation of the JNK (c-Jun N-terminal kinase) and AMPK (5’ AMP-activated protein kinase) pathways. All of these mechanisms enhance oxidative stress and lipid accumulation and promote inflammation, thereby contributing to the development of obesity and metabolic diseases, including NAFLD [30].
Mitochondrial ROS production results from dysfunction in complexes I and II of the mitochondrial respiratory chain. Various mechanisms leading to this dysfunction and ROS production have been described in NAFLD. In ob/ob mice, the quantity of mitochondrial respiratory chain complex I is decreased. In this model, mitochondrial oxidative stress causes mitochondrial DNA (mtDNA) damage, resulting in the reduction of mtDNA-encoded subunits of respiratory chain complex I. mtDNA is in the mitochondrial matrix near the mitochondrial respiratory chain where most ROS are generated. Thus, it is particularly prone to oxidative damage compared with nuclear DNA, which is more protected inside the cell nucleus. Furthermore, mtDNA lacks protective histones, which together with the absence of an mtDNA repair system, makes it more susceptible to damage[31]. Mitochondrial complex I dysfunction can also arise from mitochondrial protein 3-tyrosine nitration by peroxynitrite anion. Peroxynitrite originates from a combination of nitric oxide (NO.) with superoxide (O2.−), which are both elevated in ob/ob mice and in patients with NAFLD. In vitro, incubation of mitochondrial proteins from wild-type (WT) mice with peroxynitrite induces their 3-tyrosine nitration, which results in decreased complex I activity and mitochondrial oxygen consumption. This decrease is amplified by reduced expression of prohibitin, a protein that protects mitochondrial complexes against degradation[31]. Complex I or II activity is also regulated by the mitochondrial histone deacetylase sirtuin (SIRT) 3. Mice fed an HFD have decreased SIRT3 expression, which results in a higher level of membrane transport chain complex I acetylation, causing mitochondrial dysfunction. MCD-fed mice deficient in SIRT3 develop more severe liver lesions, inflammation, and fibrosis compared with WT mice [32]. The proper functioning of the mitochondrial respiratory chain requires ATP, and in turn, impaired ATP production leads to transport chain complex dysfunction. In NAFLD, oxidative phosphorylation (OXPHOS) and the TCA cycle are disturbed, which causes impaired ATP production leading to an alteration in mitochondrial respiration and ROS production. In patients with NASH, antioxidant capacity and ATP production decreases, which enhances hepatocellular damage and insulin resistance, promoting NAFLD progression[33].
Mitochondria are dynamic organelles, and fission and fusion are crucial for maintaining their function under environmental stress. Both processes are regulated by specific proteins, Fis-1 and Drp-1 for fission and Mfn-2 for fusion, which show decreased expression in WD-fed mice, suggesting that mitochondrial dynamism is affected in NAFLD[34]. ROS accumulation in mitochondria leads to modification of several mechanisms associated with oxidative stress and NAFLD development. Phosphorylated JNK under lipotoxicity or ER stress conditions phosphorylates SH3-domain binding protein 5 at the outer mitochondrial membrane, leading to the inactivation of mitochondrial c-Src, which regulates the phosphorylation of respiratory chain components. This Src inactivation alters electron transport, which promotes increased ROS release [35]. Phosphorylated JNK is also associated with insulin resistance, which further suggests its involvement in NAFLD development [36]. Oxidative stress induced in HFD-fed mice causes liver damage through mitochondrial membrane potential alteration [30]. ROS accumulation in mouse models of NAFLD affects mitochondrial depolarization potential through the formation of aldehydes (malondialdehyde [MDA] and 4-hydroxynonenal [4-HNE]) by lipid peroxidation or an increase in mitochondrial sensitivity to Ca2+.
Mitochondrial depolarization is a dysfunction that appears early in NAFLD development and contributes to mitochondrial homeostasis deregulation. In fact, WD-fed mice show mitochondrial depolarization at an early stage of NASH, with an increased protein level of PINK1 (PTEN-induced kinase 1), a mediator of mitochondrial autophagy (mitophagy), which is associated with mitochondrial dysfunction. In these situations of mitochondrial depolarization, the mitophagic burden increases, mitochondrial biogenesis declines, and mitochondrial depletion occurs after 2 to 6 months. These alterations are thought to be important in promoting steatosis, inflammation, and progression to fibrosis [34].
The ROS H2O2, produced by mitochondria, activates AMPK through modification of the ATP/ADP ratio [37], which further regulates antioxidant enzyme gene expression through Nrf2 activation [38]. The AMPK pathway may be activated by an antioxidant component Peroxiredoxin 5 (Prx5). Indeed, in HepG2 cells exposed to FFAs, mitochondrial Prx5 activates the AMPK pathway to regulate the activity of lipogenic enzymes (ACC, SREBP-1, FA synthase) [39]. AMPK stimulates glucose and FA oxidation by induction of CPT-1 and acyl-CoA dehydrogenase expression via PPAR gamma coactivator 1-alpha and PPARα [38]. Thus, although the AMPK pathway represents a protective response against oxidative stress and lipid accumulation, prolonged oxidative stress alters this pathway, leading to lipid accumulation and further enhanced oxidative stress. A recent study showed that the AMP kinase pathway is inhibited in NASH, leading to the activation of caspase 6-associated cell death [40]. Similarly, other antioxidant responses may have deleterious effects. Early upregulation of uncoupling protein-2 (UCP-2) during steatosis protects hepatocytes against ROS production through ATP production, whereas a further increase in UCP-2 during NASH leads to chronic ATP depletion[41].
Overall, in NAFLD, mitochondria are an important source of ROS because of alterations in complex I and II activity that initially lead to the activation of antioxidant mechanisms. However, during prolonged oxidative stress, these mechanisms participate in oxidative damage and enhance mitochondrial oxidative stress, thus contributing to the development of NAFLD (Figure 1).
The ER is an organelle connected to the nuclear membrane in eukaryotic cells and is abundant in hepatocytes because of their high metabolic activity. It has multiple functions including protein synthesis, folding, modification, and trafficking, and synthesis of lipids and steroid hormones. Alteration of ER homeostasis, i.e., ER stress, can induce oxidative stress (Figure 4). The ER stress response is defined by activation of the unfolded protein response (UPR) pathway. Prolonged ER stress increases the PKR-like ER protein kinase (PERK) and activated transcription factor (ATF) 6 pathways that both lead to ROS generation through the proapoptotic C/EBP homologous protein (CHOP), a specific protein of the ER stress response (Figure 4). CHOP is involved in oxidative stress induction in mouse models of type 2 diabetes and MCD-induced steatohepatitis [15][42]. Recently, ER stress was shown to be involved in lipogenesis and in the transition of NAFLD to NASH through caspase 2 induction[43]. ER also contributes to the regulation of calcium (Ca2+) homeostasis and particularly Ca2+ storage in the ER lumen. In animal models of diabetes and obesity, the decreased activity of the ER Ca2+ pump sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) in hepatocytes leads to ER stress and induces apoptosis [42]. Moreover, impaired Ca2+ homeostasis in the ER induces mitochondria dysfunction [15].
Other studies have provided evidence for the involvement of the Ca2+ flux in oxidative stress and ER stress induction. In Buffalo rat liver (BRL-3A) cells exposed to a high FFA concentration to mimic NAFLD, induction of oxidative stress stimulates transcription and translation of the protein calcium release-activated calcium channel protein 1 (Orai1), a plasma membrane protein involved in the Ca2+ cytosolic flux. The resulting increase in Ca2+ entry into the cell activates nuclear factor-kappa B (NF-κB) signaling, which further exacerbates the Ca2+ flux, mitochondrial impairment, and ROS generation, enhancing ER stress. Other mechanisms activated by lipid accumulation and Ca2+ homeostasis impairment in the liver also enhance ER stress in NAFLD. For instance, in a mouse model of MCD-induced steatohepatitis, increased expression and activation of liver protein kinase Cδ (PKCδ) are associated with ER stress activation. Similar results have been obtained in vitro in mouse hepatic cells cultivated in a palmitic acid-enriched MCD medium. In addition, PKCδ deficiency reduces MCD- and palmitic acid-induced ER stress activation, hepatic TG accumulation, and cell death [44]. These effects may involve CHOP, because in a human hepatic cell line (L02) exposed to palmitic acid, PKCδ deficiency reduces ER stress through decreased CHOP expression. In this model, PKCδ silencing also increases SERCA activity, which improves Ca2+ homeostasis [45]. In HFD-fed rats, increased plasma FFA is associated with increased expression of markers of oxidative stress and ER stress, as well as Orai1 and NF-κB[46]. Mitochondria and peroxisome functional alterations also can induce ER stress. For example, an imbalance in redox signaling in peroxisomes upon catalase inhibition induces FA accumulation, which leads to ROS production and ER stress [22] (Figure 4).
To summarize, ER stress induced by lipid accumulation during NAFLD promotes ER ROS production and calcium homeostasis disruption that both act to increase oxidative stress during disease progression (Figure 4). ER stress also modulates expression of proteins involved in lipid and glucose metabolism, thus contributing directly to hepatic lipid accumulation [47]. Finally, in addition to mitochondria and ER, the peroxisomes, which are important in H2O2 production by peroxisomal FA oxidation, contribute to oxidative stress in NAFLD (Figure 2) [15][22][23][48].
Patients with NAFLD present with altered expression of cytochrome P (CYP)450 enzymes, which are involved in detoxification, FA oxidation, inflammation, and oxidative stress (Figure 5)[49]. In these patients, for example, increased serum lipid peroxidation correlates with a higher level of CYP4A11. In contrast, in the hepatocyte cell line HepG2 exposed to FFA, CYP4A11 inhibition led to reduced ROS production associated with decreased lipid accumulation and reduced pro-inflammatory cytokine levels, such as tumor necrosis factor α (TNF-α), interleukin (IL)-6, and IL-1β [50]. In HFD-fed mice, CYP4A deficiency reduces hepatic ER stress, apoptosis, insulin resistance, and steatosis, suggesting a role for CYP4A in NAFLD pathogenesis. CYP2E1, which is expressed in ER, mitochondria, and cytosol, is increased in experimental models of NAFLD and in patients with NASH, resulting in ROS generation and inactivation of SOD and catalase [21][51]. CYP2E1 has a high NOX activity that promotes lipid peroxidation [51]. Furthermore, overexpression of CYP2E1 in mice is associated with severe steatohepatitis and upregulation of antioxidant enzymes (SOD2, catalase, and GPX) [52]. CYP1A1 induces ROS production by the reduction of O2 to H2O2 and O2.−. It also has a role in ω-hydroxylation of PUFAs. The expression of CYP1A1 is enhanced in oleic acid-stimulated HepG2 cells. In these same cells, CYP1A1 small interfering RNA inhibits lipid peroxidation, whereas overexpression of CYP1A1 stimulates lipid peroxidation and reduces SOD expression. Collectively, these observations unveil a regulatory role for CYP1A1 in hepatic lipid peroxidation [49].
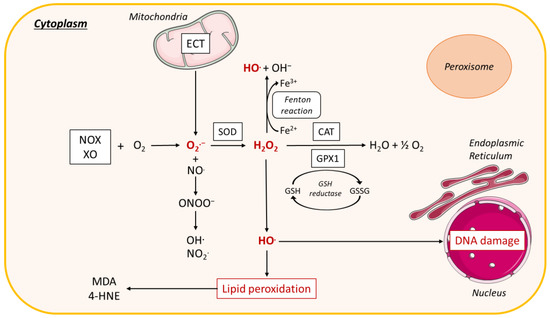
Figure 5. ROS production in the cytoplasm. In the cytoplasm, interaction of NADPH oxidase (NOX) and xanthine oxidase (XO) with oxygen (O2) leads to the formation of superoxide radical (O2.−). O2.− dismutation by the antioxidant enzyme superoxide dismutase (SOD) forms hydrogen peroxide (H2O2), which is decomposed to O2 and water (H2O) by the enzyme catalase (CAT) or the enzyme glutathione peroxidase 1 (GPX1), leading to the oxidation of reduced GSH into glutathione disulfide (GSSG). Reactive nitrogen species (RNS) are derived from nitric oxide (.NO) and superoxide (O2.−) produced via specific enzymes such as NADPH oxidase; the reaction of .NO with O2.− produces peroxynitrite (ONOO−). ONOO− can react with other molecules to form additional types of RNS including nitrogen dioxide (NO2.) as well as other types of chemically reactive free radicals (.OH). H2O2 forms a hydroxyl radical (HO.), which causes lipid peroxidation leading to malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE), or interacts with DNA causing DNA damage. H2O2 can also react with ferrous iron (Fe2+) to form hydroxyl radical (OH.) and OH−, called the Fenton reaction. Other abbreviations: NADH, nicotinamide adenine dinucleotide; H2O2, hydrogen peroxide; DNA, Deoxyribonucleic acid; XO, xanthine oxidase; ECT, electron transport chain; H2O, water.
CYP450 epoxygenase-derived epoxyeicosatrienoic acids (EETs) are a class of lipid mediators that are abundant in liver and that mediate cytoprotective and anti-inflammatory properties. A CYP450-induced EET increase in HFD-fed mice protects against NAFLD progression. Moreover, overexpression of the CYP2J2 leads to higher EET levels in serum that are associated with decreased hepatic TG levels, inhibition of the NF-κB/JNK signaling pathway, and increased antioxidant enzyme levels. Treatment of HepG2 cells with EET protects against palmitic acid-induced lipotoxicity, oxidative stress, and inflammation[53].
In cytosol, ROS are also produced by many enzymes including xanthine oxidase (XO), cyclo-oxygenase lipoxygenase, and NOX (Figure 5)[15][54][55]. NOX-mediated H2O2 production causes liver damage [56] and induces a pro-inflammatory response [54]. Patients with NAFLD have a higher NOX2 activity and increased NOX2-derived peptide, a marker of systemic NOX activation[56][57]. In one cohort of children with NAFLD, NOX2 activity increased in parallel with disease severity[56]. Furthermore, HFD-fed mice deficient in NOX2 develop steatosis but not NASH because of reduced OXPHOS dysfunction [58]. In agreement with this finding, NOX2-induced cellular oxidative stress causes inhibition of OXPHOS activity induced by FFA treatment [55]. Modulation of NOX expression also has been reported in experimental models. HFD-fed mice have increased NOX expression and activity [58], and NOX1 expression is increased in a mouse model of obesity [59], leading to ROS generation associated with nitrotyrosine protein expression that counteracted oxidative damage [60]. NOX1 also induces ROS production in a mouse model of steatosis [59].
In humans, NOX4 mRNA expression is upregulated in the liver of patients with NASH [60][61]. The NOX4 single nucleotide polymorphism rs3017887 is associated with increased ALT levels in liver biopsies from patients with NAFLD, indicating hepatocyte damage [54]. In a mouse model of diet-induced NAFLD, hepatocyte-specific deletion of NOX4 or its pharmacological inhibition reduces oxidative stress and fibrosis. Cell culture findings show that NOX4 promotes oxidative stress by ER stress activation [61]. In summary, increased NOX isoform expression and activity are associated with NAFLD and lead to oxidative stress induction via ROS generation concomitant with activation of ER and mitochondrial stress (Figure 3 and Figure 4).
Oxidized phospholipids (OxPLs) play a role in oxidative stress induction through generation of cellular ROS, which promote fibrosis and inflammation that result in NASH progression. OxPLs are produced upon overnutrition or in patients with NAFLD. Treatment of primary hepatocytes from Ldlr−/− hyperlipidemic mice with an OxPL mixture promotes ROS accumulation in the cytoplasm and all organelles, resulting in mitochondrial dysfunction[62]. Treatment of the immortalized human hepatic stellate cell line with OxPLs stimulates fibrogenic gene expression [63]. Furthermore, in a mouse model of NASH, the neutralization of OxPL decreases inflammatory mechanisms [62]. Toll-like receptor (TLR)-4 and TLR-2 are expressed in Kupffer cells and stellate cells, which are associated with fibrosis. Mice fed an HFD supplemented with lecinoxoids (synthetic OxPLs), which inhibit these TLRs, show reduced liver fibrosis and inflammation (reduced IL-1B and IL-6) [64]. Furthermore, the neutralization of OxPL in a mouse model of NAFLD inhibits progression to HCC [62].
Iron metabolism, which is modified in patients with NAFLD [65][66][67][68], leads to ROS generation because of the ability of the ferrous iron to catalyze the production of OH− from H2O2, known as the Fenton reaction [42] (Figure 2, Figure 3 and Figure 5).
Mice lacking the gut microbiota (germ-free mice) display an altered hepatic pool of glutathione[69]. The gut microbes also alter levels of amino acids and N-acetylated amino acids circulating in the mouse portal vein, affecting host amino acid and glutathione metabolism in the intestine and the liver [70]. A recent study showed that the gut microbiome induces the Nrf2 antioxidant response pathway in the liver[71] and that exogenous administration of Lactobacilli could amplify this response and protect against oxidative liver injury. Of great interest, these authors identified a Lactobacilli-derived metabolite (namely 5-methoxyindoleacetic acid) that could activate hepatic Nrf2 and in part mediate these beneficial effects. The relevance of this gut–liver mechanism in humans remains to be studied.
Unhealthy eating modifies the gut microbiota, as others have reviewed [72][73]. This dysbiosis participates in the development of metabolic diseases and plays a major role in NAFLD pathogenesis [74][75]. In fact, the development of NAFLD correlates with (i) dysbiosis; (ii) a leaky intestinal barrier; (iii) impaired mucosal immunity; (iv) bacteria and bacterial components reaching the liver through the portal vein, as well the bacterial metabolites such as lipopolysaccharides, trimethylamine-N-oxide, N,N,N-trimethyl-5-aminovaleric acid, and endogenous ethanol; and (v) impaired bile acid homeostasis [72][73]. These alterations participate in increased hepatic inflammation and oxidative stress and promote NAFLD development and progression to NASH.
Altogether, whereas the role of oxidative stress in NAFLD pathogenesis has been demonstrated in both clinical and animal studies, the underlying mechanisms are complex and not completely understood. Alterations in mitochondrial respiratory complex activity because of lipid accumulation in NAFLD make the mitochondria the main source of ROS in this condition. ER stress, cytochromes, NOX, and OxPLs are also key components in ROS generation. The consequences of ROS production in NAFLD are enhanced oxidative stress and altered lipid metabolism, which further promote lipid accumulation. In addition, increased ROS production may modulate insulin signaling and inflammatory processes, thus promoting insulin resistance and inflammation, which are important features of NAFLD progression [15] (Figure 1); however, the specific mechanisms underlying oxidative stress–promoted NAFLD need further study. Furthermore, several environmental factors, such as the nutrients and contaminants, affect oxidative stress in NAFLD.





