 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Laura Gragnani | + 3121 word(s) | 3121 | 2021-01-04 10:23:43 | | | |
| 2 | Vivi Li | Meta information modification | 3121 | 2021-01-12 03:42:21 | | |
Video Upload Options
Notch receptors are single-pass transmembrane proteins that play a critical role in cell fate decisions and have been implicated in the regulation of many developmental processes. The human Notch family comprises of four receptors (Notch 1 to 4) and five ligands. Their signaling can regulate extremely basic cellular processes such as differentiation, proliferation and death. Notch is also involved in hematopoiesis and angiogenesis, and increasing evidence suggests that these genes are involved and frequently deregulated in several human malignancies, contributing to cell autonomous activities that may be either oncogenic or tumor suppressive. It was recently proposed that Notch signaling could play an active role in promoting and sustaining a broad spectrum of lymphoid malignancies as well as mutations in Notch family members that are present in several disorders of T- and B-cells, which could be responsible for altering the related signaling. Therefore, different Notch pathway molecules could be considered as potential therapeutic targets for hematological cancers.
1. Introduction
The Notch gene was first identified in Drosophila [1] as a key developmental gene [2]. Notch receptors are single-pass transmembrane proteins which play a critical role in cell-fate decisions and have been implicated in the regulation of many developmental processes [3]. The Human Notch family comprises of four receptors (Notch 1 to 4) and five ligands— of which are members of the Delta-like (DLL1, DLL3 and DLL4) and the Jagged (JAG1 and JAG2) family [3]. Notch receptors transduce short-range signals by interacting with the transmembrane Delta-like and Jagged ligands on neighboring cells.
The Notch receptors span the cell membrane with extracellular and intracellular domains. Ligands that bind to the Notch extracellular domain result in the initiation of the sequential receptor proteolytic cleavages. In fact, an ADAM-family metalloprotease called ADAM10, cleaves the receptor just outside the membrane and the Notch extracellular domain (NECD) is released [4]. This induces γ-secretase to cleave the transmembrane region at the S3 site, releasing the Notch intracellular domain (NICD) thereby entering the cell nucleus and triggering gene expression [5]. In the nucleus, NICD forms a ternary complex with the DNA-binding protein CBF1/RBPjk/Su(H)/Lag1 (CSL) that helps recruit the adaptor protein Mastermind-like to activate target gene expression [6][7]. During the transcriptional activation process, NICD is phosphorylated on its PEST domain and targeted for proteasome-mediated degradation by ubiquitin ligases known as FBXW7. This limits the half-life of a canonical Notch signal [8].
Although Notch signaling can regulate rather basic cellular processes such as differentiation, proliferation and death, it is also involved in hematopoiesis and angiogenesis [9][10]. Increasing evidence suggests that Notch pathways are involved and frequently deregulated in several human malignancies [11], contributing to cell autonomous activities that may be either oncogenic or tumor suppressive [11].
Notch signaling plays an active role in promoting and sustaining a broad spectrum of lymphoid malignancies [12][13][14]. In addition, mutations in the Notch family members are present in several disorders of T and B-cells [11][13][15] and are responsible for altering the related signaling [12].
2. Notch 1
2.1. Physiology of Notch 1 Signaling in the Immune System Cells
Notch 1 is one of four Notch receptors expressed in mammalians. Among the five ligands, DLL4 has a higher affinity than DLL1 and JAG1 [16] and it is responsible for Notch 1 activation in the thymus of murine models [17][18]. DLL4-Notch 1 interaction is crucial in endothelial cell communication in response to vascular endothelial growth factor (VEGF) to balance tip and stalk cells in sprouting events [19].
Notch 1 is expressed in hematopoietic stem cells (HSC) and is required for their maturation, even though knockout experiments did not reveal alterations in HSC maintenance [20]. Mice with induced loss of function of Notch 1 showed blockage in T-cell development from early progenitors, before the expression of lineage surface markers [21]. In addition, Marıa J. Garcia-Leon and colleagues demonstrated that a tight regulation of Notch ligand expression in the diverse thymus region drive T-cell development [22]. In particular, the DLL4 ligand is specifically expressed in the thymus cortex epithelial cells during the embryonic phase and is downregulated in the adult thymus when the full T-cell repertoire is completed, confirming once more its pivotal role in Notch 1 induced T-cell development [22].
T-cell lineage can be distinguished into two subsets: γδ and αβ T-cells, which express different surface receptors [23]. Both T-cell subsets develop from common precursors [24]: γδ T-cells are the first to appear in the thymus during fetal development and their relative proportion decreases while αβ T-cells increase [25]. In vitro experiments with the earliest human intrathymic precursors highlighted the role of Noch1 signaling in the development of γδ T-cells [26]. Conversely, it has been shown that αβ T-cells require low levels of Notch activation [27]. Moreover, the zinc finger transcription factor Bcl11b is required for both T-cell commitment and for αβ T-cells specialization [28][29].
In fact, interaction between Notch 1 and its ligands, DLL4 and JAG2, leads γδ T-cell development through the repression of Bcl11b expression [30]. Dolens and colleagues proposed a Notch dependent temporal expression of Bcl11c. In the early critical phase of T-cell development, Notch induced the expression of the BCL11B gene while in the subsequent differentiation of γδ T-cells, induced the expression of miR-17, microRNA from the miR 17-92 cluster, which inhibits BCL11B [30]. Thus, BCL11B expression is upregulated and drives the differentiation in αβ T-cells and it is downregulated in γδ T-cells.
In addition, Ciofani et al. demonstrated that, in the thymus, the interaction between Notch 1 and Dll1 sustained pre–T-cell survival after β selection by promoting glucose metabolism through the PI3K-AKT axis [31]. Wang Q et al. reported that Notch 1 cofactor Zmiz1 is involved in the early maintenance of T-cell progenitors and in the development of T-cell lineage. Zmiz1 is probably required for normal β-selection and, in general, it is important in Notch 1-Myc-related thymocyte proliferation [32].
While the involvement of Notch 1 in T-cells has been well-investigated, its role in B-cells remains mostly unknown. Using mice models and primary mouse B-cells, Kang J.A. and collaborators showed that Notch 1 overexpression increases marginal zone B-cell numbers indicating its role in proliferation. In differentiated B-cells, Notch 1 signaling increases after the B-cell receptor binds to the antigen [33]. Notch 1 knocked-down B-cells showed a decrease in antibody production however it was reverted in splenocyte cultures where different cell types were present [34]. This suggests that Notch 1, or its ligand in other immune cells, could upregulate pathways in Notch 1-defecting B-cells [34].
Furthermore, Notch 1 target genes are involved in cell cycle, growth and survival such as c-MYC [35] and the transcription factor HES1 [36].
The role of Notch 1 in the differentiation of immune cells makes it a highly studied gene in hematologic malignancies, but it is also implicated in a wide range of solid tumors, concerning growth and progression, such as melanoma [37], intrahepatic cholangiocarcinoma [38], prostate cancer [39] and osteosarcoma [40].
2.2. Notch 1 in T-Cell Acute and Chronic Lymphoblastic Leukemia
Since Notch 1 is a key factor in T-cell development, it is clear that it has been well investigated in T-cell diseases such as acute lymphoblastic leukemia (T-ALL). The first hint of its involvement in T-ALL pathogenesis was the discovery of (7;9) translocation, a mutation that disrupts the Notch 1 gene, fusing the 3′ end portion encoding its intracellular domain (ICN) to the enhancer and promoter elements of the T-cell receptor (TCR) [41]. This results in the overexpression of a constitutively active form of Notch 1, activating genes that inhibit cell differentiation [41]. In fact, the abnormal Notch 1 signaling consequent to t(7;9) supports the growth of human translocated cell lines [42]. Less macroscopic mutations, although directly activating, were then found in the extracellular heterodimerization domain (HD) and/or the C-terminal PEST intracellular domain of the Notch 1 gene [43] and in the extracellular juxtamembrane region [44] in about 60% and 30% of T-ALL cases, respectively.
In this context, Notch 1 blocking molecules such as γ-secretase inhibitors (GSIs) could be of aid in T-ALL treatment [44] (Figure 1). γ-secretase is a protease complex that cleaves the transmembrane protein that has been studied as a target therapy for Alzheimer’s disease [45]. As Notch 1 is involved in different cancers, GSIs have been proposed as a potential drug for different solid tumors and T-ALL [46].
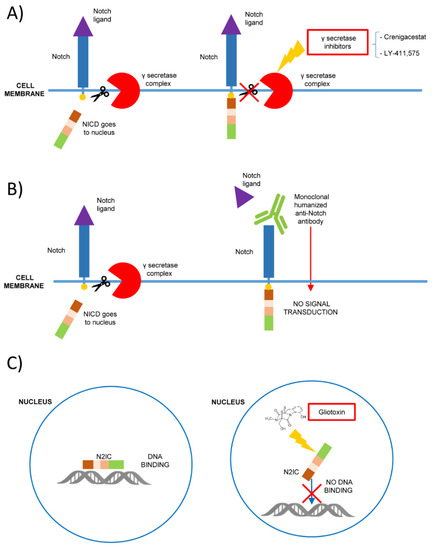
Figure 1. Mechanisms of Notch signaling inhibition as possible therapeutic strategies in hematological malignancies. (A) The inhibition of γ secretase-complex (i.e., with Crenigacestat or LY-411,575) prevents the proteolytic cleavage of Notch intracellular domain (NICD), blocking its signaling; (B) the binding with monoclonal humanized antibodies prevents Notch ligand interaction and the signaling activation; (C) gliotoxin is a specific Notch 2 inhibitor that binds Notch 2 intracellular domain (N2IC) blocking the DNA binding and preventing the target expression.
3. Notch 2
3.1. Physiology of Notch 2 Signaling in the Immune System Cells
The Notch 2 molecules are synthesized as single precursor proteins that are cleaved in the Golgi to become functional heterodimeric receptors present on the cell surface [47].
As previously explained, the interaction of Notch receptor with ligands results in further proteolytic cleavage, with the release and nuclear translocation of the NICD which functions as a transcriptional regulator [48].
NICD enters the nucleus and seeks coactivators such as Mastermind-like proteins (MAML 1-3). Together with the recombinant signal-binding protein for immunoglobulin kappa J region (RBPJ) transcription factor, they form the transcription complex accountable for the induction of Notch target genes [49].
Notch 2 is expressed in many cell types in the hemato-lymphoid compartment with specific roles in the development and differentiation of immune system cells.
In the spleen, Notch 2 activation is crucial for marginal zone (MZ) B-cells growth [50] and for the dendritic cells (DCs) differentiation [51].
Mature splenic B-cells are divided into two types of B-cells, follicular B (FOB) cells and MZB-cells [52]. Immature B-cells, developing in the bone marrow, migrate to the spleen and initially differentiate into type 1 (T1) transitional B-cells that subsequently become type 2 (T2) transitional B-cells [53]. Thereafter, these progenitors differentiate into the others two cellular types, MZB and FOB-cells. Notch 2 and no other Notch family member plays a role in the step from T2 B-cells to MZB and its activation is mediated by the DLL1 ligand [50]. DLL1 binding to the Notch 2 receptor triggers the Notch 2 signaling cascade through ADAM10-mediated cleavage and the conditionally targeted deletion of Notch 2 results in a defect of the MZB-cells [50].
Furthermore, it was recently discovered that Notch 2-dependent transcription is active in classical DCs and that it is selectively required for support of the germinal center reaction [54]. In fact, while Notch 2-dependent classical DCs are required in vivo for the induction of follicular Th cells [54], Notch 2 activation in the DC of the intestinal lamina propria corresponds to an increase of IL-17-producing CD4+ T-cells [51].
Notch 2 double deficient animals disclose differentiation of naïve CD4+ T-cells towards Th2 cells [55]. In Th1 differentiation, the role of Notch 2 signaling is less clear, as some studies showed that Th1 differentiation is increased by the Notch family members without specific details and focuses attention on the role of the two ligands Delta1 and Delta4 [56]. However, Th1 cell response is also present in Notch 2, Notch 1 and RBP-J knocked-down mice [55]. This observation leads to the hypothesis that the development of Th1 is regulated by several members of the Notch family or by all of them in a redundant way.
Maekawa Y. and co-workers [57] showed that Notch 2 signaling is able to promote cytotoxic T-cells (CTLs). In fact, in mice, Notch 2-deficient T-cells altered differentiation into CTLs and DCs with low expression of the ligand DLL1 that caused a less efficient differentiation of CTLs [57]. The intracellular domain of Notch 2 interacts with the cAMP responsive element binding protein 1 (CREB-1) [58] that binds a DNA nucleotide sequence present in many cellular promoters stimulating transcription. The control of T-cell cytotoxicity depends on the combining of Notch 2 and CREB1 signals [57].
The CTLs are involved in cancer immunosurveillance, and Notch 2, unlike Notch 1, seems to be required for the efficient induction of their antitumoral activity [59].
It is well-known that Notch signaling is involved in the self-renewal of stem cells and in the growth and differentiation of progenitors in various organs. In mice, Notch 2 improved the formation rate of short-term repopulating multipotential progenitor cells (MPPs) as well as long term repopulating HSCs [60]. Varnum-Finney B. et al. demonstrated in their studies that a Notch 2-dependent role in ensuring repopulation by HSCs, MPPs and lymphoid cells is present during BM regeneration [60]. In fact, Witt et al. previously described a double role of Notch 2 in T and B lineage stem cells, artificially inducing various levels of an activated form of Notch 2 (N2IC) in murine hematopoietic progenitors [61]. While high levels of N2IC led to T-cell leukemia, lower levels boosted the maturation of the CD8 lineage. In contrast, concerning the B lineage, N2IC drove towards the development of the B1 B-cell subset [61]. The different expression levels of N2IC suggests that Notch 2 signaling modulation may have significant consequences for thymocyte development. In mice expressing high levels of N2IC, peripheral T-cell populations have high proliferation rates. Conversely, in mice with low levels of N2IC, no disease is diagnosed until month 10 after transplantation [61]. In addition, the endogenous Notch 2 protein is more expressed in the pro-B-cells, as their development requires a crosstalk with bone marrow stroma. As Notch 2 is expressed in both MZ B and B1 B-cells, but not in FO B or B2 cells, members of the NF-κB pathway, targeted by Notch signaling, may be involved in the development of both MZ B and B1 B-cells subsets. In summary, the effect of N2IC on the B-cell lineage confirms the involvement of Notch 2 signaling in the development of B-cell subsets [61].
Considering the crucial role of Notch 2 in different aspects of the physiological development/maturation processes and homeostasis of immune system cells, it is not hard to understand how different alterations in its signaling could be involved in hematologic malignancies. These alterations are mostly somatic mutations determining gain-of-function with a potentiated Notch 2 transcriptional activity.
3.2. Notch 2 Mutations in B-Cell Lymphomas
The hyperactivation of Notch 2 through gain-of-function mutations in subtypes of mature B-cell neoplasms has been proven by Lee et al. [47]. These mutations were the partial or complete deletion of the PEST domain, or a single amino acid substitution at the C-terminus of the Notch 2 protein [47]. Notch 2 mutations were identified in DLBCL [47] and in MZB-cell lymphoma [62][63]. The PEST domain mutations cause an increased stability of Notch 2, prolonging its life. In fact, PEST sequences trigger the ubiquitin-mediated proteolysis [64] and mutations in this region, producing Notch 2 truncated forms, could prevent receptor degradation and increase its activity. The presence of recurrent somatic gain-of-function mutations in Notch 2 was also proven, using whole-genome DNA sequencing (WGS), by Kiel and colleagues [63]. They found two main types of mutations: alterations in the PEST domain and, more rarely, nonsynonymous substitutions affecting the extracellular HD [63]. Overall, only MZL (25% splenic and 5% non-splenic) showed Notch 2 mutations that were not detectable in other hematologic malignancies [63].
In addition, the authors compared mutated to wild type Notch 2 lymphomas regarding the clinical outcomes, reporting a shorter relapse-free survival, for cases carrying Notch 2 mutations [63]. Notch 2 mutations in Splenic MZL (SMZL) were identified by Rossi and colleagues as truncating events (14 frameshift indels and 11 nonsense mutations) and clustered within a hotspot region in exon 34, including a recurrent p.R2400 nonsense mutation in 24% of cases [65]. In the same study, Notch 2 mutations were described in a small percentage (3.7%) of DLBCL [65]. These observations led to a further investigation focused on this high-grade histotype [66]. The authors evaluated HCV-positive and negative DLBCL and observed a frequency of 25% in mutations in the Notch 1 and Notch 2 genes in the HCV-positive samples while only in 1% of the DLBCL negative for HCV [66]. Since these mutations were associated with a severe prognosis and with the presence of low-grade components in the diagnostic biopsy, the authors suggested that at least a fraction of the HCV-positive DLBCL could represent the transformed phase of an MZL clone or the coexistence of high and low-grade components [66]. A similar pathogenetic hypothesis was previously and cautiously suggested by Lee et al. who did not find Notch 2 mutations in MZB-cell lymphomas, but proposed the interesting possibility that some or all the DLBCL cases carrying Notch 2 mutations were transformed from MZB-cell lymphoma [47].
As already mentioned, PEST domain somatic mutations were also described for high-grade forms such as the DLBCL [47]. In this setting, a genome-wide approach revealed a rarer copy number increase of the mutated Notch 2 allele [47], a phenomenon previously associated with oncogenic gene alterations in other cancers [67].
Overall, Notch 2 mutations are involved in the pathogenic processes of MZL and DLBCL. Although the fine mechanisms still need to be understood, these alterations could represent a hypothetical biomarker and in addition the Notch 2 mutations could help inform SMZL diagnosis and prognosis [68].
3.3. Notch 2 in B-Cell Acute and Chronic Lymphocytic Leukemia
Acute myeloid leukemia (AML) is the most common myeloid malignancy in adulthood, characterized by the impairment of myeloid differentiation and blast-cell accumulation in the BM [69]. Chemo-resistance is a not rare feature of AML cells, usually due to the persistence of residual blast-cells in the BM. Signals coming from stromal cells could be responsible for inducing these kinds of abilities in AML cells through the involvement of different pathways, including Wnt/β-catenin and Notch [70].
In detail, in AML the Notch 2 pathway is boosted by receptor overexpression. Such an overexpression is not caused by a mutational event but by an altered microenvironmental signaling from bone-marrow mesenchymal stromal cells [71] inducing AML cell survival and chemo-resistance [71].
In their study on CLL, Mangolini et al. [72] used an opposite approach, based on the same concept that malignant B-cells do not survive nor proliferate autonomously, but rather in conjunction with tumorigenic cues from the stromal microenvironment. Since CLL cells constitutively express the Notch ligands JAG1, JAG2 and DLL, the authors evaluated the expression and activation of Notch 1 and Notch 2 in stromal cells [72]. They found a double role for stromal Notch 2 activation. Notch 2 activation regulates N-cadherin expression in CLL cells, interacting with and further stabilizing β-catenin and, at the same time, it boosts canonical Wnt signaling in the same CLL cells [72]. All these effects were reverted by the pharmacological inhibition of the Wnt pathway as well as the inhibition of Notch 2 in stromal cells [72].
Sera from patients with chronic B-CLL contain high amounts of soluble CD23 (sCD23), which reflects disease activity and is an important indicator of disease progression as confirmed by numerous reports [73].
CD23, which usually is a transitionally expressed marker of B-cells, is a downstream target of Notch 2 signaling [74][75]. The membrane CD23 undergoes spontaneous proteolysis, producing abnormal levels of soluble (sCD23) molecules in CLL. In this study, Hubmann et al. demonstrated a direct link between Notch 2 signaling ‘‘gain-of-function’’ and the higher levels of CD23 and in a further study, showed that peripheral CLL cells overexpressed a transcriptionally active form of Notch 2 [75]. This transcriptionally active Notch 2 truncated form acts independently from the proteolytic cleavage of γ-secretase complex that cuts the intracellular domain of the wild type Notch making it able to translocate to the nucleus. Thus, GSIs are putative novel anticancer agents at least in some Notch -associated malignancies [75]. These limitations could be overcome by a molecule selectively inhibiting all the Notch 2 isoforms, namely gliotoxin that promotes the induction of apoptosis in CLL cells contrasting the negative effects of stromal crosstalk [76][77] (Figure 1).
References
- Artavanis-Tsakonas, S.; Matsuno, K.; Fortini, M.E. Notch signaling. Science 1995, 268, 225–232.
- Fortini, M.E.; Artavanis-Tsakonas, S. Notch: Neurogenesis is only part of the picture. Cell 1993, 75, 1245–1247.
- Kovall, R.A.; Blacklow, S.C. Mechanistic Insights into Notch Receptor Signaling from Structural and Biochemical Studies. Curr. Top. Dev. Biol. 2010, 92, 31–71.
- Van Tetering, G.; Van Diest, P.; Verlaan, I.; Van Der Wall, E.; Kopan, R.; Vooijs, M. Metalloprotease ADAM10 Is Required for Notch 1 Site 2 Cleavage. J. Biol. Chem. 2009, 284, 31018–31027.
- Christian, L.M. The ADAM family. Fly 2012, 6, 30–34.
- Mumm, J.S.; Kopan, R. Notch Signaling: From the Outside In. Dev. Biol. 2000, 228, 151–165.
- Petcherski, A.G.; Kimble, J. Mastermind is a putative activator for Notch. Curr. Biol. 2000, 10, R471–R473.
- Kopan, R. Notch Signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a011213.
- Phng, L.-K.; Gerhardt, H. Angiogenesis: A Team Effort Coordinated by Notch. Dev. Cell 2009, 16, 196–208.
- Kojika, S.; Griffin, J.D. Notch receptors and hematopoiesis. Exp. Hematol. 2001, 29, 1041–1052.
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The Varied Roles of Notch in Cancer. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 245–275.
- Chiang, M.Y.; Radojcic, V.; Maillard, I. Oncogenic Notch signaling in T-cell and B-cell lymphoproliferative disorders. Curr. Opin. Hematol. 2016, 23, 362–370.
- Arruga, F.; Vaisitti, T.; Deaglio, S. The NOTCH Pathway and Its Mutations in Mature B Cell Malignancies. Front. Oncol. 2018, 8, 550.
- Pancewicz-Wojtkiewicz, J.; Nicot, C. Current views on the role of Notch signaling and the pathogenesis of human leukemia. BMC Cancer 2011, 11, 502.
- Onaindia, A.; Medeiros, L.J.; Patel, K.P. Clinical utility of recently identified diagnostic, prognostic, and predictive molecular biomarkers in mature B-cell neoplasms. Mod. Pathol. 2017, 30, 1338–1366.
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722–735.
- Koch, U.; Fiorini, E.; Benedito, R.; Besseyrias, V.; Schuster-Gossler, K.; Pierres, M.; Manley, N.R.; Duarte, A.; Macdonald, H.R.; Radtke, F. Delta-like 4 is the essential, nonredundant ligand for Notch 1 during thymic T cell lineage commitment. J. Exp. Med. 2008, 205, 2515–2523.
- Hozumi, K.; Mailhos, C.; Negishi, N.; Hirano, K.-I.; Yahata, T.; Ando, K.; Zuklys, S.; Holländer, G.A.; Shima, D.T.; Habu, S. Delta-like 4 is indispensable in thymic environment specific for T cell development. J. Exp. Med. 2008, 205, 2507–2513.
- Hellström, M.; Phng, L.-K.; Hofmann, J.J.; Wallgard, E.; Coultas, L.; Lindblom, P.; Alva, J.A.; Nilsson, A.-K.; Karlsson, L.; Gaiano, N.; et al. Dll4 signalling through Notch 1 regulates formation of tip cells during angiogenesis. Nat. Cell Biol. 2007, 445, 776–780.
- Yuan, J.S.; Kousis, P.C.; Suliman, S.; Visan, I.; Guidos, C.J. Functions of Notch Signaling in the Immune System: Consensus and Controversies. Annu. Rev. Immunol. 2010, 28, 343–365.
- Radtke, F.; Wilson, A.; Stark, G.; Bauer, M.; Van Meerwijk, J.; Macdonald, H.; Aguet, M. Deficient T Cell Fate Specification in Mice with an Induced Inactivation of Notch. Immunity 1999, 10, 547–558.
- García-León, M.J.; Fuentes, P.; De La Pompa, J.L.; Toribio, M.L. Dynamic regulation of NOTCH 1 activation and Notch ligand expression in human thymus development. Development 2018, 145, dev165597.
- Morath, A.; Schamel, W.W. αβ and γδ T cell receptors: Similar but different. J. Leukoc. Biol. 2020, 107, 1045–1055.
- Silva-Santos, B.; Pennington, D.J.; Hayday, A.C. Lymphotoxin-Mediated Regulation of Cell Differentiation by T Cell Progenitors. Science 2005, 307, 925–928.
- Chien, Y.-H.; Meyer, C.; Bonneville, M. γδT Cells: First Line of Defense and Beyond. Annu. Rev. Immunol. 2014, 32, 121–155.
- García-Peydró, M.; De Yébenes, V.G.; Toribio, M.L. Sustained Notch 1 signaling instructs the earliest human intrathymic precursors to adopt a γδ T-cell fate in fetal thymus organ culture. Blood 2003, 102, 2444–2451.
- Van De Walle, I.; De Smet, G.; De Smedt, M.; Vandekerckhove, B.; Leclercq, G.; Plum, J.; Taghon, T. An early decrease in Notch activation is required for human TCR-αβ lineage differentiation at the expense of TCR-γδ T cells. Blood 2009, 113, 2988–2998.
- Ikawa, T.; Hirose, S.; Masuda, K.; Kakugawa, K.; Satoh, R.; Shibano-Satoh, A.; Kominami, R.; Katsura, Y.; Kawamoto, H.; Pocivavsek, L.; et al. An Essential Developmental Checkpoint for Production of the T Cell Lineage. Science 2010, 329, 93–96.
- Li, L.; Leid, M.; Rothenberg, E.V. An Early T Cell Lineage Commitment Checkpoint Dependent on the Transcription Factor Bcl11b. Science 2010, 329, 89–93.
- Dolens, A.; Durinck, K.; Lavaert, M.; Van Der Meulen, J.; Velghe, I.; De Medts, J.; Weening, K.; Roels, J.; De Mulder, K.; Volders, P.; et al. Distinct Notch 1 and BCL11B requirements mediate human γδ/αβ T cell development. EMBO Rep. 2020, 21, e49006.
- Ciofani, M.; Zúñiga-Pflücker, J.C. Notch promotes survival of pre-T cells at the β-selection checkpoint by regulating cellular metabolism. Nat. Immunol. 2005, 6, 881–888.
- Wang, Q.; Yan, R.; Pinnell, N.; McCarter, A.C.; Oh, Y.; Liu, Y.; Sha, C.; Garber, N.F.; Chen, Y.; Wu, Q.; et al. Stage-specific roles for Zmiz1 in Notch -dependent steps of early T-cell development. Blood 2018, 132, 1279–1292.
- Kang, J.-A.; Kim, W.-S.; Park, S.-G. Notch 1 is an important mediator for enhancing of B-cell activation and antibody secretion by Notch ligand. Immunology 2014, 143, 550–559.
- Zhu, G.; Wang, X.; Xiao, H.; Liu, X.; Fang, Y.; Zhai, B.; Xu, R.; Han, G.; Chen, G.; Hou, C.; et al. Both Notch 1 and its ligands in B cells promote antibody production. Mol. Immunol. 2017, 91, 17–23.
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. NOTCH 1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266.
- Jarriault, S.; Le Bail, O.; Hirsinger, E.; Pourquié, O.; Logeat, F.; Strong, C.F.; Brou, C.; Seidah, N.G.; Israël, A. Delta-1 Activation of Notch -1 Signaling Results inHES-1 Transactivation. Mol. Cell. Biol. 1998, 18, 7423–7431.
- Yang, Z.; Qi, Y.; Lai, N.; Zhang, J.; Chen, Z.; Liu, M.; Zhang, W.; Luo, R.; Kang, S. Notch 1 signaling in melanoma cells promoted tumor-induced immunosuppression via upregulation of TGF-β. J. Exp. Clin. Cancer Res. 2018, 37, 1–13.
- Guo, J.; Fu, W.; Xiang, M.; Zhang, Y.; Zhou, K.; Xu, C.-R.; Li, L.; Kuang, D.; Ye, F. Notch 1 Drives the Formation and Proliferation of Intrahepatic Cholangiocarcinoma. Curr. Med. Sci. 2019, 39, 929–937.
- Rice, M.A.; Hsu, E.-C.; Aslan, M.; Ghoochani, A.; Su, A.; Stoyanova, T. Loss of Notch 1 Activity Inhibits Prostate Cancer Growth and Metastasis and Sensitizes Prostate Cancer Cells to Antiandrogen Therapies. Mol. Cancer Ther. 2019, 18, 1230–1242.
- Yu, L.; Xia, K.; Gao, T.; Chen, J.; Zhang, Z.; Sun, X.; Simões, B.M.; Eyre, R.; Fan, Z.; Guo, W.; et al. The Notch Pathway Promotes Osteosarcoma Progression through Activation of Ephrin Reverse Signaling. Mol. Cancer Res. 2019, 17, 2383–2394.
- Ellisen, L.W.; Bird, J.; West, D.C.; Soreng, A.; Reynolds, T.C.; Smith, S.D.; Sklar, J. TAN-1, the human homolog of the Drosophila Notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 1991, 66, 649–661.
- Weng, A.P.; Nam, Y.; Wolfe, M.S.; Pear, W.S.; Griffin, J.D.; Blacklow, S.C.; Aster, J.C. Growth Suppression of Pre-T Acute Lymphoblastic Leukemia Cells by Inhibition of Notch Signaling. Mol. Cell. Biol. 2003, 23, 655–664.
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris IV, J.P.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating Mutations of NOTCH 1 in Human T Cell Acute Lymphoblastic Leukemia. Science 2004, 306, 269–271.
- Sulis, M.L.; Williams, O.; Palomero, T.; Tosello, V.; Pallikuppam, S.; Real, P.; Barnes, K.; Zuurbier, L.; Meijerink, J.; Ferrando, A.A. NOTCH 1 extracellular juxtamembrane expansion mutations in T-ALL. Blood 2008, 112, 733–740.
- Kumar, D.; Ganeshpurkar, A.; Kumar, D.; Modi, G.; Gupta, S.K.; Singh, S.K. Secretase inhibitors for the treatment of Alzheimer’s disease: Long road ahead. Eur. J. Med. Chem. 2018, 148, 436–452.
- Ranganathan, P.; Weaver, K.L.; Capobianco, A.J. Notch signalling in solid tumours: A little bit of everything but not all the time. Nat. Rev. Cancer 2011, 11, 338–351.
- Lee, S.-Y.; Kumano, K.; Nakazaki, K.; Sanada, M.; Matsumoto, A.; Yamamoto, G.; Nannya, Y.; Suzuki, R.; Ota, S.; Ota, Y.; et al. Gain-of-function mutations and copy number increases of Notch 2 in diffuse large B-cell lymphoma. Cancer Sci. 2009, 100, 920–926.
- Artavanis-Tsakonas, S.; Simpson, P. Choosing a cell fate: A view from the Notch locus. Trends Genet. 1991, 7, 403–408.
- Wang, J.; Dong, M.; Xu, Z.; Song, X.; Zhang, S.; Qiao, Y.; Che, L.; Gordan, J.; Hu, K.; Liu, Y.; et al. Notch 2 controls hepatocyte-derived cholangiocarcinoma formation in mice. Oncogene 2018, 37, 3229–3242.
- Saito, T.; Chiba, S.; Ichikawa, M.; Kunisato, A.; Asai, T.; Shimizu, K.; Yamaguchi, T.; Yamamoto, G.; Seo, S.; Kumano, K.; et al. Notch 2 Is Preferentially Expressed in Mature B Cells and Indispensable for Marginal Zone B Lineage Development. Immunity 2003, 18, 675–685.
- Lewis, K.L.; Caton, M.L.; Bogunovic, M.; Greter, M.; Grajkowska, L.T.; Ng, D.; Klinakis, A.; Charo, I.F.; Jung, S.; Gommerman, J.L.; et al. Notch 2 Receptor Signaling Controls Functional Differentiation of Dendritic Cells in the Spleen and Intestine. Immunity 2011, 35, 780–791.
- Martin, F.; Kearney, J.F. Marginal-zone B cells. Nat. Rev. Immunol. 2002, 2, 323–335.
- Loder, B.F.; Mutschler, B.; Ray, R.J.; Paige, C.J.; Sideras, P.; Torres, R.; Lamers, M.C.; Carsetti, R. B Cell Development in the Spleen Takes Place in Discrete Steps and Is Determined by the Quality of B Cell Receptor–Derived Signals. J. Exp. Med. 1999, 190, 75–90.
- Briseño, C.G.; Satpathy, A.T.; Iv, J.T.D.; Ferris, S.T.; Durai, V.; Bagadia, P.; O’Connor, K.W.; Theisen, D.J.; Murphy, T.L.; Murphy, K.M. Notch 2-dependent DC2s mediate splenic germinal center responses. Proc. Natl. Acad. Sci. USA 2018, 115, 10726–10731.
- Amsen, D.; Blander, J.; Lee, G.R.; Tanigaki, K.; Honjo, T.; Flavell, R.A. Instruction of Distinct CD4 T Helper Cell Fates by Different Notch Ligands on Antigen-Presenting Cells. Cell 2004, 117, 515–526.
- Sun, J.; Krawczyk, C.J.; Pearce, E.J. Suppression of Th2 cell development by Notch ligands Delta 1 and Delta 4. J. Immunol. 2008, 180, 1655–1661.
- Maekawa, Y.; Minato, Y.; Ishifune, C.; Kurihara, T.; Kitamura, A.; Kojima, H.; Yagita, H.; Sakata-Yanagimoto, M.; Saito, T.; Taniuchi, I.; et al. Notch 2 integrates signaling by the transcription factors RBP-J and CREB1 to promote T cell cytotoxicity. Nat. Immunol. 2008, 9, 1140–1147.
- Taylor, A.K.; Klisak, I.; Mohandas, T.; Sparkes, R.S.; Li, C.; Gaynor, R.; Lusis, A.J. Assignment of the human gene for CREB1 to chromosome 2q32.3–q34. Genomics 1990, 7, 416–421.
- Sugimoto, K.; Maekawa, Y.; Kitamura, A.; Nishida, J.; Koyanagi, A.; Yagita, H.; Kojima, H.; Chiba, S.; Shimada, M.; Yasutomo, K. Notch 2 Signaling Is Required for Potent Antitumor Immunity In Vivo. J. Immunol. 2010, 184, 4673–4678.
- Varnum-Finney, B.; Halasz, L.M.; Sun, M.; Gridley, T.; Radtke, F.; Bernstein, I.D. Notch 2 governs the rate of generation of mouse long- and short-term repopulating stem cells. J. Clin. Investig. 2011, 121, 1207–1216.
- Witt, C.M.; Hurez, V.; Swindle, C.S.; Hamada, Y.; Klug, C.A. Activated Notch 2 Potentiates CD8 Lineage Maturation and Promotes the Selective Development of B1 B Cells. Mol. Cell. Biol. 2003, 23, 8637–8650.
- Trøen, G.; Wlodarska, I.; Warsame, A.; Llodrà, S.H.; De Wolf-Peeters, C.; Delabie, J. NOTCH 2 mutations in marginal zone lymphoma. Haematologica 2008, 93, 1107–1109.
- Kiel, M.J.; Velusamy, T.; Betz, B.L.; Zhao, L.; Weigelin, H.G.; Chiang, M.Y.; Huebner-Chan, D.R.; Bailey, N.G.; Yang, D.T.; Bhagat, G.; et al. Whole-genome sequencing identifies recurrent somatic NOTCH 2 mutations in splenic marginal zone lymphoma. J. Exp. Med. 2012, 209, 1553–1565.
- Rechsteiner, M.; Rogers, S.W. PEST sequences and regulation by proteolysis. Trends Biochem. Sci. 1996, 21, 267–271.
- Rossi, D.; Trifonov, V.; Fangazio, M.; Bruscaggin, A.; Rasi, S.; Spina, V.; Monti, S.; Vaisitti, T.; Arruga, F.; Famà, R.; et al. The coding genome of splenic marginal zone lymphoma: Activation of NOTCH 2 and other pathways regulating marginal zone development. J. Exp. Med. 2012, 209, 1537–1551.
- Arcaini, L.; Rossi, D.; Lucioni, M.; Nicola, M.; Bruscaggin, A.; Fiaccadori, V.; Riboni, R.; Ramponi, A.; Ferretti, V.V.; Cresta, S.; et al. The NOTCH pathway is recurrently mutated in diffuse large B-cell lymphoma associated with hepatitis C virus infection. Haematologica 2014, 100, 246–252.
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.-S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A Gain-of-Function Mutation ofJAK2in Myeloproliferative Disorders. New Engl. J. Med. 2005, 352, 1779–1790.
- Arcaini, L.; Rossi, D.; Paulli, M. Splenic marginal zone lymphoma: From genetics to management. Blood 2016, 127, 2072–2081.
- Kumar, C.C. Genetic Abnormalities and Challenges in the Treatment of Acute Myeloid Leukemia. Genes Cancer 2011, 2, 95–107.
- Kamdje, A.H.N.; Bassi, G.S.; Pacelli, L.; Malpeli, G.; Amati, E.; Nichele, I.; Pizzolo, G.; Krampera, M. Role of stromal cell-mediated Notch signaling in CLL resistance to chemotherapy. Blood Cancer J. 2012, 2, e73.
- Kamga, P.T.; Bassi, G.; Cassaro, A.; Midolo, M.; Di Trapani, M.; Gatti, A.; Carusone, R.; Resci, F.; Perbellini, O.; Gottardi, M.; et al. Notch signalling drives bone marrow stromal cell-mediated chemoresistance in acute myeloid leukemia. Oncotarget 2016, 7, 21713–21727.
- Mangolini, M.; Götte, F.; Moore, A.; Ammon, T.; Oelsner, M.; Lutzny-Geier, G.; Klein-Hitpass, L.; Williamson, J.C.; Lehner, P.J.; Dürig, J.; et al. Notch 2 controls non-autonomous Wnt-signalling in chronic lymphocytic leukaemia. Nat. Commun. 2018, 9, 1–17.
- DiRaimondo, F.; Albitar, M.; Huh, Y.; O’Brien, S.; Montillo, M.; Tedeschi, A.; Kantarjian, H.; Lerner, S.; Giustolisi, R.; Keating, M. The clinical and diagnostic relevance of CD23 expression in the chronic lymphoproliferative disease. Cancer 2002, 94, 1721–1730.
- Hubmann, R.; Schwarzmeier, J.D.; Shehata, M.; Hilgarth, M.; Duechler, M.; Dettke, M.; Berger, R. Notch 2 is involved in the overexpression of CD23 in B-cell chronic lymphocytic leukemia. Blood 2002, 99, 3742–3747.
- Hubmann, R.; Düchler, M.; Schnabl, S.; Hilgarth, M.; Demirtas, D.; Mitteregger, D.; Hölbl, A.; Vanura, K.; Le, T.; Look, T.; et al. NOTCH 2 links protein kinase C delta to the expression of CD23 in chronic lymphocytic leukaemia (CLL) cells. Br. J. Haematol. 2010, 148, 868–878.
- Hubmann, R.; Hilgarth, M.; Schnabl, S.; Ponath, E.; Reiter, M.; Demirtas, D.; Sieghart, W.; Valent, P.; Zielinski, C.; Jäger, U.; et al. Gliotoxin is a potent NOTCH 2 transactivation inhibitor and efficiently induces apoptosis in chronic lymphocytic leukaemia (CLL) cells. Br. J. Haematol. 2013, 160, 618–629.
- Hubmann, R.; Schnabl, S.; Araghi, M.; Schmidl, C.; Rendeiro, A.F.; Hilgarth, M.; Demirtas, D.; Ali, F.; Staber, P.B.; Valent, P.; et al. Targeting Nuclear NOTCH 2 by Gliotoxin Recovers a Tumor-Suppressor NOTCH 3 Activity in CLL. Cells 2020, 9, 1484.


