 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hye Won Lee | + 3534 word(s) | 3534 | 2021-01-06 03:04:11 | | | |
| 2 | Camila Xu | Meta information modification | 3534 | 2021-03-18 03:58:13 | | |
Video Upload Options
Hepatitis B virus (HBV) is a small, enveloped, and partially double-stranded DNA virus of the Hepadnaviridae family that infects hepatocytes, establishes its replication cycle, and persists in the nucleus.
1. Introduction
Hepatitis B virus (HBV) infection is a leading cause of chronic liver disease worldwide. Antiviral agents, such as interferon and nucleos(t)ide analogues (NAs), have been used to treat patients with chronic hepatitis B (CHB). Antiviral treatment strongly suppresses HBV replication and slows progression to cirrhosis and hepatocellular carcinoma (HCC). However, current antivirals are not curative, because covalently closed circular DNA (cccDNA) persists in the hepatocyte nucleus [1]. Thus, new treatment targets against HBV are needed. There are approximately 30 candidates under investigation as new treatments of HBV infection [2].
HBV is a small, enveloped, and partially double-stranded DNA virus of the Hepadnaviridae family that infects hepatocytes, establishes its replication cycle, and persists in the nucleus [3]. The HBV virion is a lipid-based spherical structure on which three viral envelope proteins (small, middle, and large) are exposed [3]. The large envelope protein, which contains the receptor-binding domain, is involved in viral entry into cytoplasm by receptor-mediated endocytosis using the sodium taurocholate co-transporting polypeptide (NTCP) receptor in the hepatocyte membrane [4][5].
The viral envelope surrounds a nucleocapsid that contains a partially double-stranded, relaxed circular DNA (rcDNA) genome [6]. In the cytoplasm of HBV-infected hepatocytes, the nucleocapsid is transported through the nuclear pore complex by microtubule-mediated transport. Following capsid dissociation via unknown factors, rcDNA is released into the nucleus and is converted to cccDNA by host factors, forming a stable mini-chromosome [7][8]. The cccDNA is transcribed into pregenomic RNA (pgRNA), which is translated to the nucleocapsid protein and the HBV DNA polymerase and serves thereafter as the template for rcDNA synthesis. The nucleocapsid with partially double-stranded HBV DNA is enveloped and the virion is secreted. A part of the nucleocapsid is recycled into the nucleus and the rcDNA is converted to cccDNA. Each step of the HBV lifecycle is a potential therapeutic target for inhibiting HBV replication and reducing HBV infectivity (e.g., perinatal infection).
2. What is the Definition of an HBV Cure?
Substantial progress has been made in the treatment of CHB in the last two decades. There are currently nine approved drugs for the treatment of CHB, including two formulations of interferon (IFN)—conventional and pegylated IFN (PegIFN)—and seven NAs: lamivudine, telbivudine, adefovir, entecavir, tenofovir disoproxil fumarate (TDF), tenofovir alafenamide fumarate (TAF), and besifovir dipivoxil (only in Korea).
The primary treatment goals for CHB are to prevent disease progression and increase survival. The therapeutic goals of current antiviral treatment are mainly virologic and biochemical responses related to the improvement of clinical outcomes [9]. A virologic response during NA therapy is defined as undetectable HBV DNA based on assays with a lower limit of detection of 10–20 IU/mL in blood. With IFN-based treatment, a virologic response is defined as a serum HBV DNA level of less than 2000 IU/mL when assessed at 6 months after the start of treatment and at the end of therapy. A biochemical response is defined as normalization of serum alanine aminotransferase. A normalization of alanine aminotransferase with a reduction of HBV viral load is an important goal to be achieved. However, these treatments do not generally promise a functional cure of CHB, which is an ideal goal of antiviral treatment.
A functional cure, defined as a sustained hepatitis B surface antigen (HBsAg) loss or seroconversion based on assays with a lower limit of HBsAg detection of 0.05 IU/mL, is a rare event in the natural history of CHB that is associated with a reduced risk of HCC [10][11]. In a population-based cohort study, the cumulative HCC incidence rate was significantly lower in participants with HBsAg seroclearance than in HBsAg-persistent carriers [12]. The HCC risk was similarly low after either spontaneous or NA-induced HBsAg seroclearance [13]. Complete cure is defined as an elimination of cccDNA together with durable HBsAg loss and undetectable serum HBV DNA. Although liver biopsy is needed to measure intrahepatic cccDNA activity, serum biomarkers that reflect cccDNA levels are needed instead. As candidates, HBV RNA, hepatitis B core-related antigen, and/or quantitative HBsAg have been investigated [14]. Quantification and ratio of large and middle proteins of HBsAg also showed specific patterns across different phases of hepatitis B that would predict the viral activity [15].
3. Targets for an HBV Cure
Several drugs are now under development that directly target the HBV replication cycle or enhance the human immune response (Figure 1). New drugs for HBV cure include agents that directly target virus life cycle or those that indirectly modulate host factor/host immune response (Table 1).
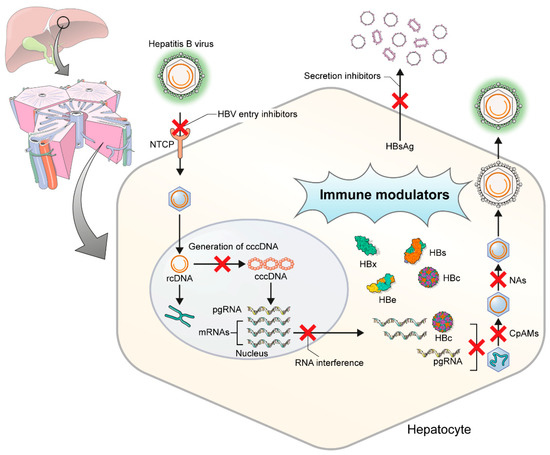
Figure 1. Targets of hepatitis B virus replication in hepatocytes. HBV, hepatitis B virus; NTCP, Na(sodium) taurocholate co-transporting polypeptide; rcDNA, relaxed circular DNA; cccDNA, covalently closed circular DNA; pgRNA, pregenomic RNA; CpAMs, core protein assembly modulators; NAs, nucleos(t)ide analogues.
Table 1. Summary of new antiviral drugs for hepatitis B cure.
| Target | Phase | Drug Name | Mode of Action | Company | Administration | NCT Number |
|---|---|---|---|---|---|---|
| Entry inhibitors | II | Bulevirtide | HBV entry inhibition | Hepateral Ltd | SC | NCT03852433 |
| Core protein allosteric modulators | I | NVR 3–778 | Assembly modulator | Novira Therapeutics, Inc. | Oral | NCT02401737 |
| II | GLS4 | Core protein binding | Sunshine Lake Pharma Co., Ltd. | Oral | NCT04147208 | |
| I | RO7049389 | Core protein binding | Hoffmann-La Roche | Oral | NCT02952924 | |
| II | JNJ-56136379 (JNJ-6379) |
Assembly modulator | Janssen, Scotland | Oral | NCT03982186 | |
| II | ABI-H0731 (Vebicorvir) | Core protein binding | Assembly Biosciences | Oral | NCT03780543 | |
| II | ABI-H2158 | Core protein binding | Assembly Biosciences | Oral | NCT04398134 | |
| 1 | JNJ-64530440 (JNJ-0440) |
Assembly modulator | Alios Biopharma Inc. | Oral | NCT03439488 | |
| II | QL-007 | Assembly modulator | Qilu Pharmaceutical Co., Ltd. | Oral | NCT04157699 | |
| RNA interference | II | ARC-520 | RNA interference | Arrowhead Pharmaceuticals | IV | NCT02577029 |
| II | INOIS-HBVRx (GSK3228836) |
Antisense oligonucleotides | GlaxoSmithKline | SC | NCT02981602 | |
| Preclinical | INOIS-HBVLRx (GSK33389404) |
Antisense oligonucleotides | GlaxoSmithKline | - | - | |
| II | VIR-2218 | RNA interference | Vir Biotechnology, Inc. | SC | NCT04412863 | |
| II | ARO-HBV (JNJ-3989) | RNA interference | Arrowhead Pharmaceuticals | SC | NCT03365947 | |
| I | DCR-HBVS | RNA interference | Dicerna Pharmaceuticals, Inc. | SC | NCT03772249 | |
| Inhibition of HBsAg release | II | REP 2139-Ca | Inhibition of HBsAg release | Replicor Inc. | IV | NCT02726789 |
| II | REP 2139-Mg | Inhibition of HBsAg release | Replicor Inc. | IV | NCT02565719 | |
| HBsAg neutralization | II | GC 1102 (Lenvervimab) | Neutralization and inhibiting reentry | Green Cross Corporation | IV | NCT03801798 |
| Inhibitors of cccDNA | Preclinical | TALENs | cccDNA disruption | - | - | - |
| Preclinical | CRISPR-Cas9 | cccDNA disruption | - | - | - | |
| Toll-like receptor agonists | II | GS-9620 (vesatolimod) | TLR7 agonist | Gilead Sciences | Oral | NCT02166047 |
| II | GS-9688 (selgantolimod) | TLR8 agonist | Gilead, USA | Oral | NCT03615066 | |
| I | RO7020531 | TLR7 agonist | Hoffmann-La Roche | Oral | NCT02956850 | |
| Immune checkpoint inhibitors | I | Nivolumab | Anti-PD-1 | PharmaEssentia | IV | NCT04638439 |
| I/II | REGN2810 (cemiplimab) | Anti-PD-1 | Regeneron Pharmaceuticals | IV | NCT04046107 | |
| Therapeutic vaccines | I | INO-1800 | DNA plasmids | Inovio Pharmaceuticals | E-IM | NCT02431312 |
| I | TG1050 | HBV proteins | Transgene | SC | NCT02428400 | |
| I | ChAdOx1 HBV | Adjuvanted ChAd and MVA vectored | Vaccitech Limited | IM | NCT04297917 | |
| I | JNJ-64300535 | DNA vaccines | Janssen Sciences Ireland UC | E-IM | NCT03463369 | |
| II | GS-4774 | DNA vaccines | Gilead Sciences | SC | NCT02174276 |
Abbreviations: HBV, hepatitis B virus; SC, subcutaneous; IV, intravenous; HBsAg, hepatitis B surface antigen; cccDNA, covalently closed circular DNA; TALENs, transcription activator-like effector nuclease; TLR, toll-like receptor; Anti-PD-1, anti-programmed cell death protein 1; IM, intramuscular; E-IM electroporation-mediated intramuscular. References: https://www.clinicaltrials.gov (updated on 20 December 2020).
3.1. Direct-Acting Antivirals
3.1.1. Entry Inhibitors
Bulevirtide (formerly Myrcludex B) is a synthetic N-acetylated lipopeptide that contains the NTCP-binding pre-S1 domain of the large HBsAg envelope protein. A nanomolar range of affinity makes bulevirtide competitively bind to NTCP and block de novo HBV infection by competing with viral pre-S1 motif to bind with NTCP [16][17]. Bulevirtide was recently approved in the European Union for the treatment of chronic hepatitis delta virus (HDV) in HDV RNA-positive patients [18]. In a phase IIa study, 40 hepatitis B e antigen (HBeAg)-negative patients were randomized to receive bulevirtide at one of five dosages (0.5, 1, 2, or 5 mg for 12 weeks, and 10 mg once daily for 24 weeks). Bulevirtide was administered by subcutaneous injection. A decline in HBV greater than 1 log was observed in six out of eight patients in the 10 mg dosing group and in seven out of twenty-one patients in the lower dosing groups. However, no patient achieved HBsAg loss [19]. To date, most studies of bulevirtide have focused on chronic HDV infection. Sixty patients with HBV/HDV coinfection were randomized to receive PegIFN alone, PegIFN plus bulevirtide 2 or 5 mg, or bulevirtide 2 mg for 48 weeks [20]. Among the 60 patients, 20% of patients in the PegIFN plus bulevirtide 2 mg group achieved HBsAg loss at 24 weeks off therapy, and 27% achieved HBsAg loss at 48 weeks off therapy. In recent data (NCT03546621), 30 patients were randomized to PegIFN plus bulevirtide 10 mg once per day for 48 weeks, or TDF plus bulevirtide 5 mg twice a day for 48 weeks, together with sequential TDF for up to 72 weeks. At week 72, a higher proportion of the bulevirtide 5 mg plus TDF group showed undetectable HDV RNA, but HBsAg was undetectable in a lower proportion. The two groups showed the same proportions of alanine aminotransferase (ALT) normalization. There were no serious treatment-related adverse events or discontinuation of treatment. Most of these studies have been applied to HBV/HDV coinfection, but we expect a potent synergistic effect on patients with CHB when entry inhibitors and other antivirals are combined.
3.1.2. Core Protein Allosteric Modulators
HBV core protein is essential for HBV pgRNA packaging and reverse transcription. Several compounds referred to as core protein allosteric modulators (CpAMs, also known as capsid assembly modulators in the literature) are under investigation. Depending on the chemical structure of CpAMs, unstable aberrant capsids or empty capsids can be formed. Two classes of CpAMs have been identified based on their mechanism of action [21]. Class I CpAMs, typified by heteroaryl dihyropyridines, increase the kinetics of capsid formation and lead to the formation of misassembled capsids. Class II CpAMs are typified by phenylpropenamides, which accelerate capsid assembly and form morphologically normal capsids that are empty and lack viral pgRNA and HBV polymerase.
NVR 3–778 was the first-in-class CpAM evaluated either alone or in combination with PegIFN. In a phase I study of HBeAg-positive patients with CHB without cirrhosis, reductions in both HBV DNA and HBV RNA were greater in the group with NVR 3–778 plus PegIFN than in the group with NVR 3–778 or PegIFN alone [22]. However, viral rebound was observed after drug cessation.
JNJ-6379 (JNJ-56136379) is a class II CpAM that binds to the HBV core protein and disrupts early- and late-stage processes in the HBV life cycle [14]. In a phase I study of treatment-naïve patients with CHB, a 4-week administration of JNJ-6379 was tolerated, showed dose-dependent pharmacokinetics, and had potent antiviral activity (decreases in HBV DNA and HBV RNA) [23]. Primarily, JNJ-6379 interferes with capsid assembly kinetics, preventing encapsidation of pgRNA and blocking HBV replication (late step in the viral life cycle). In addition, JNJ-6379 can also inhibit de novo formation of cccDNA, potentially interfering with the capsid disassembly process. JNJ-6379 blocks production of Dane particles and RNA-containing particles. However, since it does not target HBsAg, subviral particles of HBsAg can still be released to the blood. A phase II study is ongoing.
RO7049389 is an oral small molecule and class I HBV CpAM that induces formation of abnormal HBV core aggregates. In a recent phase I study, RO7049389 was administered to HBV patients at different dose regimens for 28 days and resulted in a robust decline in median HBV DNA (2.7 to 3.0 log10 IU/mL), which is promising for virus suppression. Viral rebound was observed after stopping treatment [24].
ABI-H0731 (vebicorvir) is a potent and selective class II CpAM. Thirty-eight non-cirrhotic patients were randomly assigned to receive ABI-H0731 or placebo and treated once daily for up to 28 days in a double-blind, placebo-controlled study. In the second study, vebicorvir 300 mg plus entecavir or entecavir plus placebo daily was given to treatment-naïve HBeAg-positive patients [25]. Compared to entecavir plus placebo, the combination of vebicorvir plus entecavir resulted in significant declines at week 12 in both HBV DNA and HBV RNA. In recent data (NCT03576066), 100% of HBeAg-negative CHB patients (n = 18) had virologically suppressed HBV DNA plus pgRNA levels <20 IU/mL after 48 weeks of treatment with vebicorvir 300 mg once daily in combination with oral entecavir, TDF or TAF. Based on the possibilities of using capsid inhibitors plus NAs to substantially decrease HBV DNA levels, a phase III study is in preparation. Long-term studies are needed to determine whether CpAMs can eliminate HBsAg, HBeAg and/or cccDNA.
3.1.3. RNA Interference
RNA interference (RNAi) can target HBV transcripts directly and induce their degradation. RNAi is a highly specific and efficient method of post-transcriptional gene silencing [26]. The synthetic small interfering RNA (siRNA) interferes with the expression of a specific target gene by degrading mRNA. Unlike NA therapy, siRNA shuts down HBsAg production and may lead to the restoration of the immune response through rapid HBsAg reduction and breakdown of immune tolerance. A major limitation of this drug is the issue of delivery, and the lack of a reduction in cccDNA. Antisense oligonucleotides are small nucleic acids that are complementary to the target transcript and act through steric hindrance and/or RNA degradation by ribonuclease H cleavage.
ARC-520 was investigated in two randomized, multicenter studies of NA-experienced patients who were HBeAg-negative or HBeAg-positive [27]. HBsAg levels were significantly reduced in the high-dose groups (2 mg/kg ARC-520 combined with NAs) compared to a placebo group, with mean reductions of 0.38 log IU/mL in HBeAg-negative patients and 0.54 log IU/mL in HBeAg-positive patients; the reductions persisted for approximately 85 days and >85 days after the last dose. A modified RNAi, JNJ-3989 (formerly ARO-HBV), contains two RNAi that are both conjugated to N-acetyl galactosamine to facilitate uptake by the liver. ARO-HBV induced rapid declines in HBV DNA and HBsAg in patients with CHB [28]. In recent data, JNJ-3989 injections with entecavir or TDF resulted in maintained HBsAg declines ≥1 log10 IU/mL in 39% (15/38) of patients with CHB.
An alternative approach to blocking viral protein expression is to use liver-directed antisense oligonucleotides. IONIS-HBVRx (GSK3228836) and IONIS-HBVLRx (GSK33389404) allow delivery of antisense molecules to the liver via the hepatocyte-expressed asialoglycoprotein. This approach may reduce off-target toxicities associated with antisense oligonucleotides [29].
3.1.4. Inhibition of HBsAg Release
The most abundant viral antigen in the blood is HBsAg, which plays an important role in preventing immune control of HBV [8][30]. Circulating HBsAg is almost entirely in the form of non-infectious HBV subviral particles (SVPs) [31][32][33]. Because these particles are produced independently of viral replication, the viral antigen is difficult to target using the therapies approved to date.
Nucleic acid polymers, such as REP 2139, block the release of HBsAg from infective hepatocytes by selectively targeting the assembly and/or secretion of SVPs [34]. REP 2139 naturally enters liver cells (hepatocytes), where it prevents the assembly of SVPs in any hepatocyte producing these particles. This mechanism effectively hinders the replenishment of HBsAg in the blood and reduces HBsAg levels within hepatocytes. The overall antiviral effect of REP 2139 allows the body to clear HBsAg and thereby reduce or remove the inhibition of immune control caused by this viral antigen. The current formulation of REP 2139 (REP 2139-Mg) induces few to no side effects and is typically administered once every week for 48 weeks by intravenous infusion in combination with other antiviral agents. Moreover, REP 2139-Mg is expected to be equally effective with a once-weekly injection under the skin (subcutaneous injection), a regimen that will be used in future trials.
Recently, the safety and efficacy of the addition of either REP 2139-Mg or REP 2165-Mg to a backbone of TDF and PegIFN was investigated in an open-label, randomized phase II study of HBeAg-negative CHB patients [35]. After the 48-week treatment, HBsAg levels were ≤0.05 IU/mL in 60% (24/40) of the patients. During a further 48-week of treatment-free follow-up, virologic control persisted in 32.5% (13/40) of patients. Among patients with persistent HBsAg seroconversion, functional cure was maintained in 35% (14/40). Thus, the administration of HBsAg release inhibitors together with immune modulators may be an effective combination regimen.
3.1.5. Neutralization
Hepabig gene (lenvervimab) is a recombinant human monoclonal antibody that binds to HBsAg to neutralize HBV. This compound not only inhibits viral entry into hepatocytes but also enhances the immune response by significantly reducing HBsAg levels. The mechanisms involve the neutralization of circulating virion or surface antigen by the formation of immune complexes and the inhibition of viral re-entry by binding to HBsAg. In a prospective, open-label phase I trial, single and multiple doses of lenvervimab were administered in four different doses (80,000 IU, 120,000 IU, 180,000 IU or 240,000 IU) to HBeAg-positive patients, and virological suppression occurred in HBeAg-negative CHB patients naturally or with NA therapy [36]. Lenvervimab showed good tolerability as well as a correlation between the baseline HBsAg level and sustained HBsAg loss. It is now undergoing a double-blind, randomized, phase IIa study to evaluate its efficacy and safety when administered in combination with NAs.
3.1.6. Inhibitors of cccDNA
cccDNA serves as the template for viral transcription, and pgRNA as the template for viral replication. Thus, disabling cccDNA can be an effective curative option for HBV infection. Numerous small molecules have been developed as sequence-specific RNA-guided nucleases and proteins that can putatively block the formation, enhance the destruction, and silence the transcription of cccDNA while stimulating cell division [37]. These include a cleaving sequence-specific DNA targets using the transcription activator-like (TAL) effector nucleases (TALENs) or a gene-editing using the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated 9 (Cas9) system, showing antiviral efficacy [38][39]. Epigenetic modification by histone modification and cccDNA methylation can modify actively transcribed DNA to an inactive status without changing the DNA itself [40]. Potential inhibitors of cccDNA such as directly gene-editors, epigenetic modifiers and DNA destabilizer are on development, however, they have yet to be evaluated in clinical trials.
3.2. Immune Modulatory Therapies or Indirect Antivirals
A weak innate and HBV-specific immunologic response occurs in patients with CHB [21]. Chronic HBV infection leads to T-cell dysfunction (“exhaustion”), a state characterized by poor effector cytotoxic activity, impaired cytokine production, and the sustained expression of multiple inhibitory receptors [41]. Thus, the identification of immunomodulatory targets is important in therapeutic strategies aimed at restoring HBV-specific immune responses with immunomodulatory agents. However, the results achieved thus far with immune modulators have been disappointing.
3.2.1. Toll-Like Receptor Agonists
Toll-like receptors (TLRs) are the initial sensors of viral infection and initiate the intracellular pathways that induce the production of antiviral mediators [42]. The activation of TLR-mediated pathways results in suppression of HBV replication and restoration of HBV-specific adaptive immunity. In a double-blind, randomized, placebo-controlled phase II study, patients received once-weekly oral vesatolimod (GS-0620, TLR-7 agonist) or placebo [43]. Vesatolimod induced an HBV-specific immune response, but without a significant decline in HBsAg. Selgantolimod (GS-9688, TLR-8 agonist) is now in a phase II study. Selgantolimod (1.5 mg or 3 mg once a day) was administered with NA therapy for 24 weeks, followed by NA therapy alone for 24 weeks. HBsAg loss was achieved in 5% (2/39) of the patients, and in 16% (3/19) of patients who were HBeAg-positive. Another study (NCT03615066) that tested selgantolimod plus TAF against a placebo plus TAF in viremic patients with CHB showed that selgantolimod is safe and well tolerated, with a decline in HBsAg levels to ≥0.3 log10 IU/mL in the selgantolimod plus TAF group.
3.2.2. Engineered T Cells
The adoptive transfer of newly engineered HBV-specific T cells may be a novel strategy [44]. Genetic reprogramming to create functional T cells to eliminate HBV-infected hepatocytes could be achieved through T-cell receptor (TCR) gene transfer or through the use of chimeric antigen receptor (CAR) T cells [21]. Tests of HBV-specific T cells with CAR or classic TCRs both in vitro and in HBV transgenic mice have shown selective elimination of HBV-infected cell lines and control of HBV replication with only transient liver damage, respectively [45][46].
3.2.3. Immune Checkpoint Inhibitors
The efficacy of targeting checkpoint inhibitors, such as programmed cell death protein 1 (PD-1) and programmed death ligand 1 (PD-L1), has been demonstrated by the restoration of vigorous immune responses in patients with malignancies. However, there are concerns about the induction of autoimmunity or hepatitis flare via nonspecific activation of the immune system.
Anti-PD-L1 may be a therapeutic candidate in patients with CHB, as it is expected to restore antiviral T-cell functions [47]. In vivo blockade of the PD-1/PD-L1 pathway in CD8 T cells, in combination with entecavir treatment and DNA vaccination, was shown to enhance the function of virus-specific T cells [48]. A phase I pilot study evaluated anti-PD-1 (nivolumab) treatment with or without GS-4774 (therapeutic vaccine) in HBeAg-negative CHB patients. At week 24, 14% (3/22) of the patients had > 0.5 log10 reduction in HBsAg levels [49].
3.2.4. Therapeutic Vaccine
The aim of therapeutic vaccines is to stimulate the host immune response to restore HBV-specific immune control while suppressing HBV replication and ultimately inducing HBsAg loss. However, these vaccines have been largely unsuccessful [50]. The only finding of efficacy was a reduction in HBV DNA levels at the end of follow-up in patients given a therapeutic vaccine plus the standard of care compared to those who received the standard of care alone. In contrast to the failure of previous vaccines that only target HBsAg, the development of newer DNA vaccines is attempting various prime-boost approaches.
Other DNA vaccines are currently under development, including heterologous prime-boost approaches, vaccines against multiple HBV proteins, and novel adjuvants [51]. INO-1800 is a vaccine consisting of a mixture of recombinant DNA encoding the HBsAg and the consensus sequence of the hepatitis B core/capsid protein antigen. INO-1800, alone or in combination with interleukin-12, is currently being investigated in patients with CHB (NCT02431312). Another candidate is JNJ-64300535, which is being tested in combination with NAs. Vaccines such as GS-4774 and TG-1050 are based on approaches using multiple HBV proteins, but they have yielded disappointing results in clinical trials. Nonetheless, preliminary data imply that therapeutic vaccines will be effective when administered in a combination approach.
References
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis, B. Gut 2015, 64, 1972–1984.
- Liang, T.J.; Block, T.M.; McMahon, B.J.; Ghany, M.G.; Urban, S.; Guo, J.T.; Locarnini, S.; Zoulim, F.; Chang, K.M.; Lok, A.S. Present and future therapies of hepatitis B: From discovery to cure. Hepatology 2015, 62, 1893–1908.
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479, 672–686.
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. elife 2012, 1, e00049.
- Blanchet, M.; Sureau, C. Infectivity determinants of the hepatitis B virus pre-S domain are confined to the N-terminal 75 amino acid residues. J. Virol. 2007, 81, 5841–5849.
- Summers, J.; O’Connell, A.; Millman, I. Genome of hepatitis B virus: Restriction enzyme cleavage and structure of DNA extracted from Dane particles. Proc. Natl. Acad. Sci. USA 1975, 72, 4597–4601.
- Kann, M.; Schmitz, A.; Rabe, B. Intracellular transport of hepatitis B virus. World J.Gastroenterol. WJG 2007, 13, 39.
- Jiang, B.; Hildt, E. Intracellular Trafficking of HBV Particles. Cells 2020, 9, 2023.
- Naggie, S.; Lok, A.S. New Therapeutics for Hepatitis B: The Road to Cure. Ann. Rev. Med. 2020, 72.
- Cornberg, M.; Lok, A.S.F.; Terrault, N.A.; Zoulim, F.; Berg, T.; Brunetto, M.R.; Buchholz, S.; Buti, M.; Chan, H.L.; The 2019 EASL-AASLD HBV Treatment Endpoints Conference Faculty; et al. Guidance for Design and Endpoints of Clinical Trials in Chronic Hepatitis B—Report From the 2019 EASL-AASLD HBV Treatment Endpoints Conference. Hepatology 2020, 71, 1070–1092.
- Tout, I.; Loureiro, D.; Mansouri, A.; Soumelis, V.; Boyer, N.; Asselah, T. Hepatitis B surface antigen seroclearance: Immune mechanisms, clinical impact, importance for drug development. J. Hepatol. 2020, 73, 409–422.
- Song, C.; Zhu, J.; Ge, Z.; Yu, C.; Tian, T.; Wang, H.; Han, J.; Shen, H.; Dai, J.; Lu, J.; et al. Spontaneous Seroclearance of Hepatitis B Surface Antigen and Risk of Hepatocellular Carcinoma. Clin. Gastroenterol. Hepatol. 2019, 17, 1204–1206.
- Yip, T.C.; Wong, V.W.; Tse, Y.K.; Liang, L.Y.; Hui, V.W.; Zhang, X.; Li, G.L.; Lui, G.C.Y.; Chan, H.L.Y.; Wong, G.L.H. Similarly low risk of hepatocellular carcinoma after either spontaneous or nucleos(t)ide analogue-induced hepatitis B surface antigen loss. Aliment. Pharmacol. Ther. 2020, 53, 321–331.
- Asselah, T.; Loureiro, D.; Boyer, N.; Mansouri, A. Targets and future direct-acting antiviral approaches to achieve hepatitis B virus cure. Lancet Gastroenterol. Hepatol. 2019, 4, 883–892.
- Pfefferkorn, M.; Böhm, S.; Schott, T.; Deichsel, D.; Bremer, C.M.; Schröder, K.; Gerlich, W.H.; Glebe, D.; Berg, T.; van Bömmel, T. Quantification of large and middle proteins of hepatitis B virus surface antigen (HBsAg) as a novel tool for the identification of inactive HBV carriers. Gut 2018, 67, 2045–2053.
- Urban, S.; Bartenschlager, R.; Kubitz, R.; Zoulim, F. Strategies to inhibit entry of HBV and HDV into hepatocytes. Gastroenterology 2014, 147, 48–64.
- Asselah, T.; Loureiro, D.; Tout, I.; Castelnau, C.; Boyer, N.; Marcellin, P.; Mansouri, A. Future treatments for hepatitis delta virus infection. Liver Int. 2020, 40 (Suppl. 1), 54–60.
- Kang, C.; Syed, Y.Y. Bulevirtide: First Approval. Drugs 2020, 80, 1601–1605.
- Bogomolov, P.; Alexandrov, A.; Voronkova, N.; Macievich, M.; Kokina, K.; Petrachenkova, M.; Lehr, T.; Lempp, F.A.; Wedemeyer, H.; Haag, M.; et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: First results of a phase Ib/IIa study. J. Hepatol. 2016, 65, 490–498.
- Wedemeyer, H.; Schoneweis, K.; Bogomolov, P.O.; Voronkova, N.; Chulanov, V.; Stepanova, T.; Bremer, B.; Allweiss, L.; Dandri, M.; Burhenne, J.; et al. Final results of a multicenter, open-label phase 2 clinical trial (MYR203) to assess safety and efficacy of myrcludex B in cwith PEG-interferon Alpha 2a in patients with chronic HBV/HDV co-infection. J. Hepatol. 2019, 70, 81.
- Spyrou, E.; Smith, C.I.; Ghany, M.G. Hepatitis B: Current Status of Therapy and Future Therapies. Gastroenterol. Clin. N. Am. 2020, 49, 215–238.
- Yuen, M.F.; Gane, E.J.; Kim, D.J.; Weilert, F.; Yuen Chan, H.L.; Lalezari, J.; Hwang, S.G.; Nguyen, T.; Flores, O.; Hartman, G.; et al. Antiviral Activity, Safety, and Pharmacokinetics of Capsid Assembly Modulator NVR 3–778 in Patients with Chronic HBV Infection. Gastroenterology 2019, 156, 1392–1403.e7.
- Zoulim, F.; Lenz, O.; Vandenbossche, J.J.; Talloen, W.; Verbinnen, T.; Moscalu, I.; Streinu-Cercel, A.; Bourgeois, S.; Buti, M.; Crespo, J.; et al. JNJ-56136379, an HBV Capsid Assembly Modulator, Is Well-Tolerated and Has Antiviral Activity in a Phase 1 Study of Patients With Chronic Infection. Gastroenterology 2020, 159, 521–533.e9.
- Yuen, M.-F.; Schwabe, C.; Tanwandee, T.; Jin, Y.; Gao, L.; Zhou, X.; Das, S.; Wang, Y.; Lemenuel-Diot, A.; Cosson, V.; et al. RO7049389, a core protein allosteric modulator, demonstrates robust decline in HBV DNA and HBV RNA in chronic HBV infected patients. Sci. HBV Cure 2019.
- Ma, X.; Lalezari, J.; Nguyen, T.; Bae, H.; Schiff, E.R.; Fung, S.; Yuen, R.M.F.; Hassanein, T.; Hann, H.W.; Elkhashab, M.; et al. LBO-06-Interim safety and efficacy results of the ABI-H0731 phase 2a program exploring the combination of ABI-H0731 with Nuc therapy in treatment-naive and treatment-suppressed chronic hepatitis B patients. J. Hepatol. 2019, 70 (Suppl. 1), e130.
- Nayagam, J.S.; Cargill, Z.C.; Agarwal, K. The Role of RNA Interference in Functional Cure Strategies for Chronic Hepatitis, B. Curr. Hepatol. Rep. 2020, 19, 362–369.
- Yuen, M.F.; Schiefke, I.; Yoon, J.H.; Ahn, S.H.; Heo, J.; Kim, J.H.; Lik Yuen Chan, H.; Yoon, K.T.; Klinker, H.; Manns, M.; et al. RNA Interference Therapy With ARC-520 Results in Prolonged Hepatitis B Surface Antigen Response in Patients With Chronic Hepatitis B Infection. Hepatology 2020, 72, 19–31.
- Gane, E.J.; Locarnini, S.; Lim, T.H.; Strasser, S.; Sievert, W.; Cheng, W.; Thompson, A.; Given, B.; Schluep, T.; Hamilton, J.; et al. First results with rna interference (rnai) in chronic hepatitis b (chb) using ARO-HBV. Hepatology 2018, 68, 1463A.
- Han, K.; Cremer, J.; Elston, R.; Oliver, S.; Baptiste-Brown, S.; Chen, S.; Gardiner, D.; Davies, M.; Saunders, J.; Hamatake, R.; et al. A Randomized, Double-Blind, Placebo-Controlled, First-Time-in-Human Study to Assess the Safety, Tolerability, and Pharmacokinetics of Single and Multiple Ascending Doses of GSK3389404 in Healthy Subjects. Clin. Pharmacol. Drug Dev. 2019, 8, 790–801.
- Real, C.I.; Werner, M.; Paul, A.; Gerken, G.; Schlaak, J.F.; Vaillant, A.; Broering, R. Nucleic acid-based polymers effective against hepatitis B Virus infection in patients don’t harbor immunostimulatory properties in primary isolated liver cells. Sci. Rep. 2017, 7, 43838.
- Franke, C.; Matschl, U.; Bruns, M. Enzymatic treatment of duck hepatitis B virus: Topology of the surface proteins for virions and noninfectious subviral particles. Virology 2007, 359, 126–136.
- Chai, N.; Chang, H.E.; Nicolas, E.; Han, Z.; Jarnik, M.; Taylor, J. Properties of subviral particles of hepatitis B virus. J. Virol. 2008, 82, 7812–7817.
- Hu, J.; Liu, K. Complete and Incomplete Hepatitis B Virus Particles: Formation, Function, and Application. Viruses 2017, 9, 56.
- Bazinet, M.; Pântea, V.; Cebotarescu, V.; Cojuhari, L.; Jimbei, P.; Albrecht, J.; Schmid, P.; Le Gal, F.; Gordien, E.; Krawczyk, A.; et al. Safety and efficacy of REP 2139 and pegylated interferon alfa-2a for treatment-naive patients with chronic hepatitis B virus and hepatitis D virus co-infection (REP 301 and REP 301-LTF): A non-randomised, open-label, phase 2 trial. Lancet Gastroenterol. Hepatol. 2017, 2, 877–889.
- Bazinet, M.; Pântea, V.; Placinta, G.; Moscalu, I.; Cebotarescu, V.; Cojuhari, L.; Jimbei, P.; Iarovoi, L.; Smesnoi, V.; Musteata, T.; et al. Safety and Efficacy of 48 Weeks REP 2139 or REP 2165, Tenofovir Disoproxil, and Pegylated Interferon Alfa-2a in Patients With Chronic HBV Infection Naïve to Nucleos(t)ide Therapy. Gastroenterology 2020, 158, 2180–2194.
- Lee, H.W.; Park, J.Y.; Hong, T.; Park, M.S.; Ahn, S.H. Efficacy of Lenvervimab, a Recombinant Human Immunoglobulin, in Treatment of Chronic Hepatitis B Virus Infection. Clin. Gastroenterol. Hepatol. 2020, 18, 3043–3045.e1.
- Ruiz de Galarreta, M.; Lujambio, A. Therapeutic editing of hepatocyte genome in vivo. J. Hepatol. 2017, 67, 818–828.
- Liu, X.; Hao, R.; Chen, S.; Guo, D.; Chen, Y. Inhibition of hepatitis B virus by the CRISPR/Cas9 system via targeting the conserved regions of the viral genome. J. Gen. Virol. 2015, 96, 2252–2261.
- Chen, J.; Zhang, W.; Lin, J.; Wang, F.; Wu, M.; Chen, C.; Zheng, Y.; Peng, X.; Li, J.; Yuan, Z.; et al. An efficient antiviral strategy for targeting hepatitis B virus genome using transcription activator-like effector nucleases. Mol. Ther. 2014, 22, 303–311.
- Hong, X.; Kim, E.S.; Guo, H. Epigenetic regulation of hepatitis B virus covalently closed circular DNA: Implications for epigenetic therapy against chronic hepatitis, B. Hepatology 2017, 66, 2066–2077.
- Ye, B.; Liu, X.; Li, X.; Kong, H.; Tian, L.; Chen, Y. T-cell exhaustion in chronic hepatitis B infection: Current knowledge and clinical significance. Cell Death Dis. 2015, 6, e1694.
- Ma, Z.; Cao, Q.; Xiong, Y.; Zhang, E.; Lu, M. Interaction between Hepatitis B Virus and Toll-Like Receptors: Current Status and Potential Therapeutic Use for Chronic Hepatitis, B. Vaccines 2018, 6, 6.
- Janssen, H.L.; Brunetto, M.R.; Kim, Y.J.; Ferrari, C.; Massetto, B.; Nguyen, A.H.; Joshi, A.; Woo, J.; Lau, A.H.; Gaggar, A.; et al. Safety, efficacy and pharmacodynamics of vesatolimod (GS-9620) in virally suppressed patients with chronic hepatitis, B. J. Hepatol. 2018, 68, 431–440.
- Bertoletti, A.; Le Bert, N. Immunotherapy for Chronic Hepatitis B Virus Infection. Gut Liver 2018, 12, 497–507.
- Gehring, A.J.; Xue, S.A.; Ho, Z.Z.; Teoh, D.; Ruedl, C.; Chia, A.; Koh, S.; Lim, S.G.; Maini, M.K.; Stauss, H.; et al. Engineering virus-specific T cells that target HBV infected hepatocytes and hepatocellular carcinoma cell lines. J. Hepatol. 2011, 55, 103–110.
- Krebs, K.; Böttinger, N.; Huang, L.R.; Chmielewski, M.; Arzberger, S.; Gasteiger, G.; Jäger, C.; Schmitt, E.; Bohne, F.; Aichler, M.; et al. T cells expressing a chimeric antigen receptor that binds hepatitis B virus envelope proteins control virus replication in mice. Gastroenterology 2013, 145, 456–465.
- Fisicaro, P.; Valdatta, C.; Massari, M.; Loggi, E.; Biasini, E.; Sacchelli, L.; Cavallo, M.C.; Silini, E.M.; Andreone, P.; Missale, G.; et al. Antiviral intrahepatic T-cell responses can be restored by blocking programmed death-1 pathway in chronic hepatitis, B. Gastroenterology 2010, 138, 682–693.
- Liu, J.; Zhang, E.; Ma, Z.; Wu, W.; Kosinska, A.; Zhang, X.; Möller, I.; Seiz, P.; Glebe, D.; Wang, B.; et al. Enhancing virus-specific immunity in vivo by combining therapeutic vaccination and PD-L1 blockade in chronic hepadnaviral infection. PLoS Pathog. 2014, 10, e1003856.
- Gane, E.; Verdon, D.J.; Brooks, A.E.; Gaggar, A.; Nguyen, A.H.; Subramanian, G.M.; Schwabe, C.; Dunbar, P.R. Anti-PD-1 blockade with nivolumab with and without therapeutic vaccination for virally suppressed chronic hepatitis B: A pilot study. J. Hepatol. 2019, 71, 900–907.
- Lim, S.G.; Agcaoili, J.; De Souza, N.N.A.; Chan, E. Therapeutic vaccination for chronic hepatitis B: A systematic review and meta-analysis. J Viral. Hepat. 2019, 26, 803–817.
- Gehring, A.J.; Protzer, U. Targeting Innate and Adaptive Immune Responses to Cure Chronic HBV Infection. Gastroenterology 2019, 156, 325–337.


