1000/1000
Hot
Most Recent
 +1 point
+1 point
The human microbiome has been identified as a potentially significant contributor to human health, but the composition and role that the microbiome plays varies across body sites. Technological improvements have enabled large-scale studies of the human microbiome and this review focuses on the data and health impacts that have been analyzed to date associated with the skin, oral, gut, and vaginal microbiomes. One major challenge in this area that remains is to gain a better mechanistic understanding of the microbial consortium function and dynamics and how it impacts the human host. Computational approaches can help analyze some of the interactions and complexity of the microbiome-host interactions and thus, discussion on relevant computational studies associated with the human microbiome are also presented.
Interest in microorganisms associated with the human body has a long history dating back to the handcrafted microscopes built by Antonie van Leewenhoek in the 1670s, where bacteria in van Leeuwenhoek’s oral and fecal samples were referred to as “animalcules” [[1]]. As research techniques and knowledge increased, human-associated microorganisms continued to be studied (e.g., use of Hungate tubes to isolate anaerobic microbes [[2]] and documented (e.g., book titled “A Flora and Fauna within Living Animals” in 1853). The accumulation of research has led to the current concept of the human microbiome that has been described as an ecological community of commensal, symbiotic and pathogenic microorganisms that literally share our body space and have been all but ignored as determinants of health and disease [[3]].
Early glimpses into the complexity of the human microbiome started in the 1980s where diverse microorganisms such as S. aureus, E. coli and Viridans were found by Rotimi et al. in the umbilical, oral and fecal flora of neonates [[4]]. More recently, Pasolli and co-workers examined the microbiomes at the four human anatomical sites: stool, vagina, skin and oral, using large-scale metagenomic approaches and found ~150,000 microbial genomes coming from 4930 species (~total number of species in the human microbiome) [[5]]. Interestingly, 77% (3796) of the identified species were novel species whose genomes were not present in any of the public repositories. The scope and novelty of composition was further emphasized where it was noted that ~75% genes associated with the human microbiome lack functional annotation meaning that there is a large amount of “functional dark matter” associated with the human microbiome [[6]]. Apart from genes, proteins and metabolites in the human microbiome, recently, researchers have found that human microbiome is not restricted to only microbes and their metabolites, genes and enzymes. Small proteins or microproteins (less than 50AA) have been regularly found in human microbiome with the possibility of millions of small open reading frames (ORFs) or microproteins being present [[7]]. The possible functions of the microproteins include: housekeeping, bacterial adaptation, bacterial defense against phages and microbe–microbe and microbe–host cell communication. These types of novel findings indicate the mysterious side of the human microbiome. Ursell et al. and Gilbert et al. have already suggested that human microbiome is still highly unexplored [[8][9]].
16S rRNA sequencing has been the standard and regular approach to find the species composition of the human microbiome [[10]]. The hypervariable regions V1–V3 and V3–V5 of the 16S rRNA gene help in identifying the taxonomic composition of various bacterial species. People have clustered this gene into operational taxonomic units (OTUs) to investigate the microbiota composition in healthy humans [[11]]. Sanger sequencing has been the standard method to sequence the complete stretch of the amplicon (16S rDNA) [[12]]. However, people realized that species composition can be identified using shorter DNA stretches with higher sequence coverage and thus next generation sequencing (NGS) technologies, i.e., Roche 454 pyrosequencing, Illumina and Ion Torrent sequencing [[12]] are also used for meta-genomic sequencing. Later, many computational approaches have also been developed to analyze the 16S rRNA sequences of both disease and non-disease causing microbes to better understand their biology in the human microbial communities [[10],[13]]. However, even if we have good coverage and longer sequencing reads using 16S rRNA sequencing, it will always be hard to obtain the genomic information of low abundance species [[6],[10]]. Therefore, recent research has shifted to using high-throughput data techniques to produce both the qualitative and quantitative knowledge of the DNA, mRNA transcripts, metabolites, and proteins of the microbial groups in the microbiome [[14][15]]. Meta-omic approaches can help provide a more comprehensive functional view of microorganisms and their roles within the microbiome. Shotgun metagenomic sequencing was the first step in this direction where bacteria’s whole genomic DNA from human/environmental samples is analyzed for both species identification and understanding gene function potential of the microbe [[12],[16]]. Another example is the HMP Unified Metabolic Analysis Network (HUMAnN) that performs metabolic and functional reconstructions of metagenomic data [[17]]. This method was applied on seven primary human body sites including stool, tongue dorsum and anterior nares on 102 individuals. They identified the core metabolic pathways, genes and functional modules which were different for different sites across individuals. In the vaginal microbiome, it was found that glycosaminoglycan degradation, phosphate and amino acid transport are more active in this microbiome [[18]].
Building upon the increased experimental data generated through the high-throughput approaches, computational modeling approaches such as genome scale metabolic models (GEMs) have been developed to integrate and analyze data to study function (Figure 1). In recent years, meta-omics data have been used in conjunction with genome scale metabolic models (GEMs). This is illustrated in Table 1, where omics and meta-omics data were used in the majority of the GEM studies. Genome scale metabolic models and metagenomics data is taken as an input when you are using MAMBO (Metabolomic Analysis of Metagenomes using fBa and Optimization) [[19]]. This research study used this approach where they incorporated 1500 microbes in their model and showed that a distinct metabolome exists at vagina, stool, skin and oral sites in the human body [[19]]. Use of in vitro, ex vivo and in vivo experimental data with in silico models serve as the excellent research pipeline to discover the unknown microbe–microbe and microbe-host metabolic interactions in human microbiomes suggesting crucial therapeutic advancements [[14]]. While each of the respective omic data types provide useful information in characterizing organism function, some of the data types are more directly converted to the modeling formalism than others. For example, Vanee et al. used a proteomics derived model to understand the metabolism functionalities of the microbe Thermobifida fusca where the growth rates shown by experimental and in silico data were almost similar [[20]]. If experimental and computational work is properly used collaboratively, there will be identification of not only the representative species of the human microbiome but of the other unknown species with whom these leader microbes coordinate through vast number of metabolite exchanges [[21]].
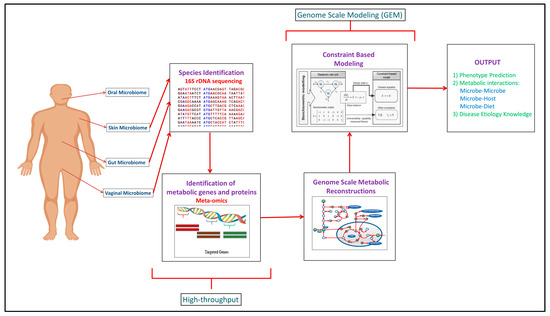
Figure 1. Complete human microbiome research pipeline established over the years explaining the integration of experimental and computational methodologies to get the mechanistic understanding of the human microbiome.
Computational analyses such as network analysis, agent-based modeling, and genome scale metabolic modeling (GEM) have used to study various aspects of the human microbiome including structure, dynamics, and coordinated function [[22][23][24]]. While a variety of specific analyses are encompassed in network analysis, generally this approach considers connectivity of components to consider structure-function relationships. This type of analysis often leads to the identification of critical, highly connected hubs/nodes and can provide insight into robustness to perturbations. Agent-based modeling is a stochastic simulation approach where discrete agents are ascribed attributes to represent a biological entity and allowed to interact dynamically with other agents. For example, agents could be used to depict an enzyme and a substrate and different concentrations of these could be simulated to study the time-course dynamics by changing binding affinities. Thus, system function can be studied by modifying properties of an agent and running simulations to see effects. Genome scale metabolic modeling is a modeling approach that formulates a model based upon experimentally-established information such as gene content (genomic data) and biochemistry, and a number of publications and reviews exist describing this method in detail [[25]]. By starting with gene content and connecting associated enzymes with biochemical function, a stoichiometric matrix is generated that allows simulations to be run that can be used to study a variety of biological systems including human–microbe and microbe–microbe interactions [[26][27]]. A number of software packages have been developed to support development and analysis of genome scale metabolic models including the constraint based reconstruction and analysis (COBRA) Toolbox [[28]]. Reconstructed networks for an organism represent the biochemical and genetic capabilities and can be analyzed by stipulating input constraints to create a space of allowable flux distributions to elucidate all of the possible metabolic flux states. Flux balance analysis (FBA) is the generic approach for calculating the flux/flow of metabolites through the network in an organism and hence, predicting the growth phenotype of the organism or rate of production of an important chemical compound [[29]]. It finds the optimal solutions to the objective function which basically depicts the biological function which the network is performing. For example, if you want to predict growth, biomass production by the target organism is taken as the objective function [[30]]. After predictions are made using FBA, validation with experimental data or model reconciliation is achieved between experimental and computational models using various algorithms [[31][32][33]]. This entire process of building, analyzing, and testing GEMs helps obtain a broad functional understanding of an organism and subsequently helps analyze and predict difference between organisms, for example, between pathogens and non-pathogens of gut microbiome to find therapeutic targets [[34]].
Given that the human microbiome involves numerous interacting species, community genome scale metabolic models are definitely required for capturing the microbiome biology in a comprehensive manner [[26],[28],[35]]. However, community genome scale modeling has challenges that need to be addressed. First, most of these studies have been done on individual species because the number of species of the human microbiome which have models is very low (~25) [[21],[28],[35]]. In the biological world, there are of course not one or two, but multiple species working either in a cooperative or competitive way in the complex microbiome. This brings us to the second challenge which is specifying a global objective function for the whole community of microbes. People, however, have constantly made efforts to tackle this problem by designing constraint based/genome scale modeling software packages. A python software tool called Micom was developed which can take into consideration the objective functions for both individual species and the microbial community including ~100 species at a time [[36]]. Baldini et al. have created a MATLAB based software called Microbiome Modeling Toolbox which applies genome scale modeling to simulate pairwise microbe–microbe and human–microbe interactions [[27]]. Compartmentalization is the third challenge where reactions and the metabolites in microbiome are needed to be partitioned correctly in the appropriate compartments (species or cell organelles) [[6],[35]].
There is microbial diversity across each of the human microbiome sites (gut, oral, skin and vagina) and temporal diversity meaning varied biogeography dynamics exist for each of these sites which advocates a more directed research towards personalized medicine for combating the human microbial diseases [[8],[9],[37]]. The microbial behaviors and the molecular mechanisms at these different human anatomical areas is different from each other [[5],[8],[9]]. The vast and complicated nature of the human microbiome (the microbiome between two individuals is 80%–90% different compared to the human genome which is 99.9% similar among individuals [[8]]) has even given rise to a new multidisciplinary field of systems microbial medicine where experts from a wide range of fields like microbiology, genetics, mathematics, statistics, engineering, computational biology, nutrition, immunology, neurology and endocrinology are required to work together to get insights to human microbiome [[38]]. The Human Microbiome Project (HMP) was a great initiative in this direction where experts from different areas are trying to investigate different aspects of human microbiome like species composition, metabolome, microbe–microbe interactions [[39]].
Studying the human microbiome is a challenging endeavor, but the connection to human health is becoming clearer as the four major human microbiomes (skin, oral, gut, and vaginal) have their own associated diseases: Crohn’s disease and obesity in gut, bacterial vaginosis and preterm birth in vaginal, periodontitis in oral, atopic dermatitis in skin [[40][41][42][43][44]]. Therefore, there is a huge need to study the human microbiome at these four anatomical sites in depth. Our review covers the background, high throughput studies, and modeling methodologies employed to study each of the four distinct microbiomes (gut, oral, skin and vaginal). Bringing all of them under one banner will help us to get a holistic view of the global human microbial interactions which can be used in future for the development of effective and novel treatment strategies.





