 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Micaela Gliozzi | + 1781 word(s) | 1781 | 2020-12-09 08:51:51 | | | |
| 2 | Vicky Zhou | Meta information modification | 1781 | 2020-12-22 06:53:03 | | |
Video Upload Options
Any damage to the endothelial cells leads to the interruption of the barrier, an increased permeability, and an inflow of molecules that generate inflammatory response. Pulmonary endothelium is able to generate bioactive molecules and/or to use compounds present in the cell to reduce the effects of toxic stimuli and restore its conditions. If the damage is particularly extensive, the endothelium cannot tackle it and its permeability undergoes some alterations. The pulmonary endothelial barrier is completely destroyed in the case of chronic lung damages. A typical example of chronic inflammation of the alveolus-capillary unit is represented by virus infections.
1. Introduction
Observational studies carried out in the development of coronavirus disease 2019 (Covid-19) pandemic revealed that the cardiovascular system represents one of the major targets of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-Cov-2) infection. In fact, it has been found that nearly 30% of SARS-CoV-2 patients undergo cardiac injury and that cardiovascular complications including acute cardiac injury, stroke, heart failure, arrhythmias, and cardiomyopathies may be detected at any stage of the infection [1]. The prevalence of cardiovascular co-morbidities, such as diabetes, hypertension, or coronary artery disease, is often associated with an unfavourable prognosis of SARS-CoV-2 infection [2]. Clinical studies in patients with SARS-Cov-2, both in China and in Italy, showed a higher mortality rate (CFR) in patients with cardiovascular co-morbidity than those without co-comorbidity [3][4][5]. Furthermore, a recent clinical study of 44,672 SARS-Cov-2-infected patients showed the CFR increased from 0.9% (in the absence of co-morbidity) to 10.5% in the presence of cardiovascular disorders to 7.3% in the presence of diabetes and to 6% in the presence of hypertension [6]. The pathophysiological mechanisms underlying the higher incidence of cardiovascular complications detected in patients undergoing severe SARS-Cov-2 disease are still to be better clarified. However, the common pathophysiological link between SARS-CoV-2 infection and the cardiovascular events leading to irreversible multi-organ dysfunction is represented by the disruption of factors that maintain the blood vessels into an anti-thrombotic state. In fact, it is possible to highlight an association between SARS-CoV-2 infection and increased risk of thromboembolism [7].
In general, thrombotic response involves two important aspects: (1) platelet activation and (2) a coagulation cascade [8].
(1) At the moment of platelet activation, arachidonic acid is converted to thromboxane A2, which is a potent pro-aggregatory and vasoconstrictive factor [9]. Then the platelets degranulate and finally undergo a conformational change assuming a starry shape, which is characteristic of their physical aggregation [10].
(2) The activation of the coagulation cascade determines, through several pathways, the formation of thrombin that splits the fibrinogen into fibrin. This fibrillar protein polymerizes to form a “mesh” together with platelets at the wound site.
It is important to point out that there is an interaction between the coagulation cascade and the activation of the platelets. In fact, thrombin can activate platelets but also conversely platelets themselves are able to catalyze the formation of thrombin [11].
During an inflammatory process that activates leukocytes, with the recruitment and translocation of other leukocytes as well as the release of cytokines and other inflammatory mediators, activation of the complement and attempt to eliminate agents have infected or attacked cells [12]. Platelets play an important role in the regulation of the inflammatory process. During acute inflammation, there is an increase in platelet-monocyte and platelet-neutrophil aggregates. In addition, platelet inhibition reduces cytokines such as Interleukin 6 (IL-6) and Tumor Necrosis Factor alpha (TNF-α) [13].
Multiple mechanisms have been suggested to approach this phenomenon. In particular, evidence exists that SARS-CoV-2 infection produces an aggressive and systemic pro-inflammatory response in which viral infection is associated with a consistent release of mediators of inflammation known as the so-called “cytokine storm” [14][15]. This leads to impressive damage of lung tissue, which becomes unable to ensure the due oxygen exchange in pulmonary tissue. Moreover, SARS-CoV-2 infection-induced hypoxia is accompanied by an enhanced tendency to develop thrombosis of blood micro-vessels via an increased blood viscosity [16]. This is also related to the general status of hospitalized patients, who are more than 60 years old, bedridden for a long time, and subjected to invasive treatments. These are risk factors to develop hyper-coagulation or thrombosis [17].
The common pathophysiological feature, which can be found in patients with irreversible cardio-respiratory complications occurring in SARS-Cov-2 disease is represented by an imbalanced anti-coagulant activity of vascular endothelial cells, which is also called “sepsis-induced coagulopathy” (SIC). This is an effect that leads to an increased risk of thrombotic events [18]. For this reason, although the correlation between SARS-CoV-2 infection and coagulation disorders is still unclear, a tight monitoring of coagulation biomarkers was suggested in high-risk patients [19].
SARS-CoV-2 infection associated coagulopathies are characterized by high levels of D-dimer (a fibrin degradation product detectable in the blood in case of fibrinolysis), fibrinogen, prothrombin time (PT), and thrombocytopenia [20]. Furthermore, this condition is accompanied by a micro-thrombosis, systemic inflammatory response, and impairment of vascular reactivity [21].
Finally, a systemic impairment of endothelial function seems to occur in the majority of patients undergoing SARS-CoV-2 infection, thereby, playing a crucial role in the SARS-CoV-2-linked sepsis-related coagulopathy [22]. In particular, emerging evidence suggests that SARS-CoV-2 could damage the endothelial barriers and this event could contribute to the severe and systemic condition generated by pandemic infection.
2. Endothelium Dysfunction and SARS-CoV-2
The flu virus infection is associated with an infiltration of white blood cells in the lungs. Neutrophils, in particular, release cytokines, reactive oxygen species, elastases, and nucleic acids that contribute to the destruction of the endothelial barrier. Moreover, cytokines and other inflammatory mediators impair the endothelial cell-cell junctions with the following formation of inter-cellular interstices, causing the loss of vascular fluid in the intermediate space, resulting in oedemas. The mechanisms underlying the junctional destruction include the reduction of Cadherin 5, type 2 (VE cadherin), which is a protein that ensures the cohesion of the endothelial cells, and the cytoskeletal rearrangement of the actin filaments [23].
Considering the previous assumptions concerning the endothelium, it would be interesting to imagine a model of the impairment of the alveolus-lung unit resulting from the virus infection caused by SARS-CoV-2. In fact, the cells of the pulmonary endothelium express the ACE2 receptor [24], and it would be argued that the virus, after having attacked and destroyed the pulmonary epithelium, could transmigrate to the underlying endothelium and penetrate the cells [25]. Thus, through the mechanisms already explained, SARS-CoV-2 could damage the endothelium and contribute to the severe and systemic condition generated by this pandemic infection.
A very interesting recent scientific work has shown the involvement of endothelial cells across vascular beds of different organs in a series of patients with SARS-CoV-2. The results obtained are related to people with SARS-CoV-2, with previous diseases, that, following the infection, died. A subsequent autopsy was carried out. Post-mortem analysis revealed the presence of viral elements within the endothelium and an accumulation of inflammatory cells that led to the death of endothelial cells. In addition, histopathological analysis showed that SARS-Cov-2 infection led to the induction of endothelitis in different organs. Finally, apoptosis and pyroptosis of endothelial cells were also highlighted [26].
Under normal conditions, the endothelium is anticoagulated by a series of natural anticoagulant systems. In case of damaged endothelium, there is a cellular alteration which, in many cases, leads to an inflammatory state known as “endotheliopathy.” In this altered condition, a dysfunction in the microcirculation occurs [27]. Endothelitis and SARS-CoV-2 inclusions were found in the vascular endothelial cells of patients who died as a result of infection and underwent autopsy. These findings have indicated alteration of endothelial cells as a central feature of the pathophysiology of SARS-CoV-2 during the inflammatory phase of the disease [28]. The concrete proof of this deduction was provided by a biochemical measurement of specific markers of endotheliopathy, haemostatic factors, and platelet activation, which were always altered in the coagulopathy associated with SARS-CoV-2 infection [29]. The Von Willebrand factor (vWF) is a large multimeric glycoprotein that performs two critical functions in primary hemostasis. It acts as a bridge molecule in vascular lesion sites for normal platelet adhesion and, under high shear conditions, promotes platelet aggregation. Factor VIII (FVIII) is an essential blood-clotting protein, also known as an anti-hemophilic factor (AHF). vWF together with AHF are common in people with SARS-CoV-2 and their levels were found to be much higher than for unaffected patients [30].
Post mortem examination of 21 people diagnosed with SARS-CoV-2 has shown that the primary cause of death was respiratory failure with massive capillary congestion and severe changes of rheological properties in capillaries. Nevertheless, subsequent findings have highlighted pulmonary embolisms (in four patients), alveolar haemorrhage (in three patients), thrombotic microangiopathy (in three patients), and vasculitis (in one patient). The observation of all patients also highlighted damage of the endothelial in the kidneys and intestines, suggesting vascular dysfunction in disease progression [31]. Another important study, carried out with an autoptic examination of 26 people who died as a consequence of SARS-CoV-2 infection, demonstrated kidney injuries with glomerular and vascular changes, occlusion of the microvascular lumens, and numerous erythrocytes as a consequence of endothelial damage [32]. The damage to the endothelium and the formation of a microthrombus, caused by SARS-CoV-2 infection, are represented in Figure 2.
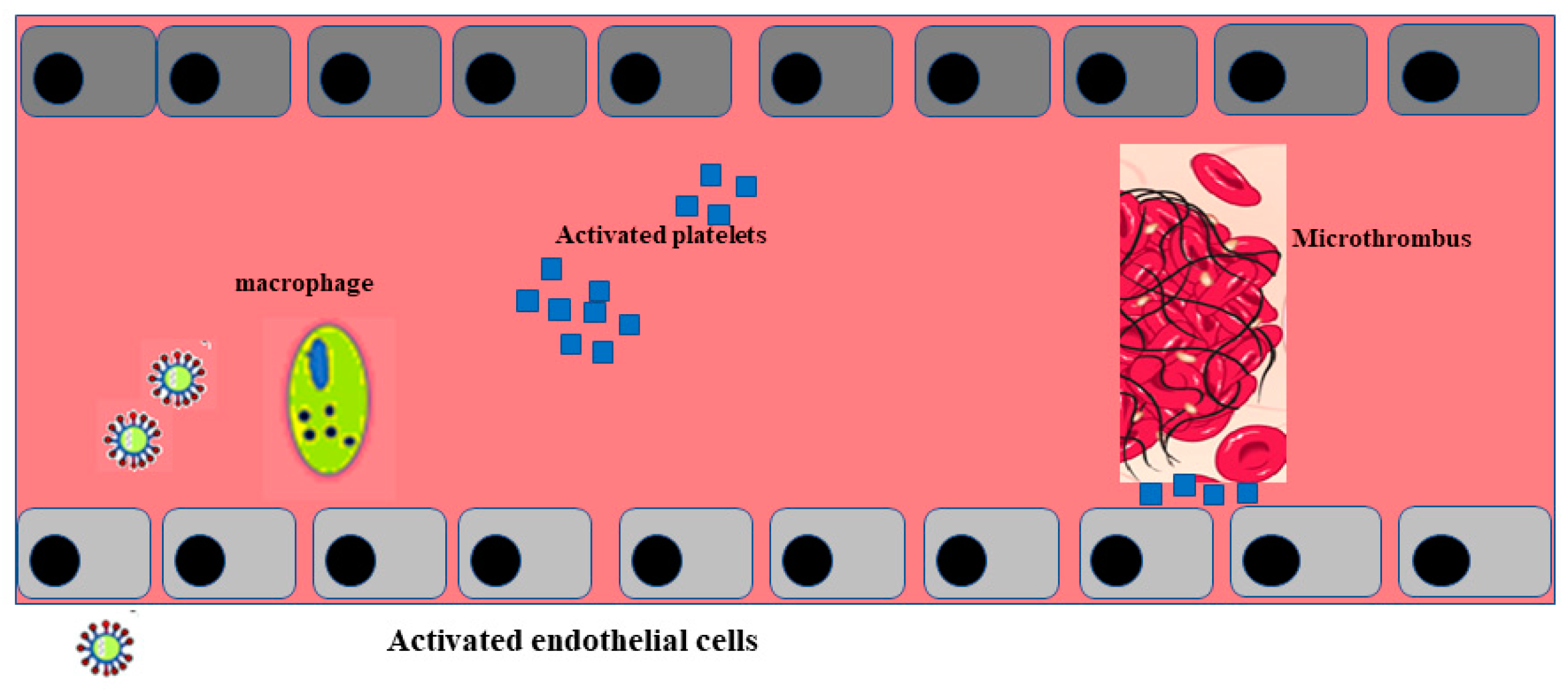
3. Conclusions
Similarities between SARS-Cov and SARS-CoV-2 are not only related to symptoms developed during infection or to the receptor mechanism of viral penetration. In fact, a common feature to both infections is to develop vascular thrombosis [33][34]. Abnormalities in the coagulation response, observed in SARS-CoV-2 infection, are not directly linked to an intrinsic characteristic of the virus but to its ability to trigger an inflammatory cascade [35]. Data reported directly from China, showed that 6% of 99 hospitalized patients affected by SARS-CoV-2 had a high prothrombin time along with a 36% of elevated D-dimer and increased biomarkers of inflammation [36]. Following a viral infection, a physiological inflammatory response is activated, which involves an alteration in the coagulation process. These events are involved in a process known as “thrombo-inflammation” or “immuno-thrombosis” [37][38].
In most patients who died of severe SARS-CoV-2 infection and who were autopsied, widespread thrombosis, and micro-vascularization were reported. Since thrombosis has also been observed in patients undergoing anticoagulant therapy, it is likely to believe that clotting disorders may be a characterizing factor of this infection [39].
According to this result, endothelial dysfunction represents a key mechanism that leads to early impairment of endogenous anti-inflammatory/anti-thrombotic responses of the vascular wall able to counteract systemic tissue damage subsequent to SARS CoV-2 infection. Moreover, the tendency to develop thrombotic vascular events associated with a SARS CoV-2 disease state must be taken into account when approaching people with SARS-CoV-2 even those who are asymptomatic. This should be crucial in preventing and treating SARS CoV-2–related systemic damage to reduce its impact in terms of hospitalization and death, which remains an unresolved issue in the course of the disease.
References
- Akhmerov, A.; Marban, E. COVID-19 and the heart. Circ. Res. 2020, 126, 14431–14455.
- Li, B.; Yang, J.; Zhao, F.; Zhi, L.; Wang, X.; Liu, L.; Bi, Z.; Zhao, Y. Prevalence and impact of cardiovascular metabolic diseases on COVID-19 in China. Clin. Res. Cardiol. 2020, 109, 5315–5338.
- Wu, Z.; McGoogan, J.M. Characteristics of and important lessonsfrom the coronavirus disease 2019 (COVID-19) outbreak in China: Summary of a report of 72 314 cases from the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242.
- Guzik, T.J.; Mohiddin, S.A.; Dimarco, A.; Patel, V.; Savvatis, K.; Marelli-Berg, F.M.; Madhur, M.S.; Tomaszewski, M.; Maffia, P.; D’Acquisto, F. COVID-19 and the cardiovascular system: Implications for risk assessment, diagnosis, and treatment options. Cardiovasc. Res. 2020, 116, 16661–16687.
- Grasselli, G.; Zangrillo, A.; Zanella, A.; Antonelli, M.; Cabrini, L.; Castelli, A.; Cereda, D.; Coluccello, A.; Foti, G.; Fumagalli, R.; et al. Baseline characteristics and outcomes of 1591 patients infected With SARS-CoV-2 admitted to ICUs of the Lombardy Region, Italy. JAMA 2020, 323, 1574–1581.
- Kunal, S.; Gupta, K.; Sharma, S.M.; Pathak, V.; Mittal, S.; Tarke, C. Cardiovascular system and COVID-19: Perspectives from a developing country. Monaldi Arch. Chest Dis. 2020, 90, 90.
- Al-Ani, F.; Chehade, S.; Lazo-Langner, A. Thrombosis risk associated with COVID-19 infection. A scoping review. Thromb. Res. 2020, 192, 152–160.
- Gąsecka, A.; Borovac, J.A.; Guerreiro, R.A.; Giustozzi, M.; Parker, W.; Caldeira, D.; Chiva-Blanch, G. Thrombotic Complications in Patients with COVID-19: Pathophysiological Mechanisms, Diagnosis, and Treatment. Cardiovasc. Drugs Ther. 2020, 1–15.
- Parker, W.; Orme, R.C.; Hanson, J.; Stokes, H.M.; Bridge, C.M.; Shaw, P.A.; Sumaya, W.; Thorneycroft, K.; Petrucci, G.; Porro, B.; et al. Very-low-dose twice-daily aspirin maintains platelet inhibition and improves haemostasis during dual-antiplatelet therapy for acute coronary syndrome. Platelets 2019, 30, 148–157.
- Paul, B.Z.; Jin, J.; Kunapuli, S.P. Molecular mechanism of thromboxane A(2)-induced platelet aggregation. Essential role for p2t(ac) and alpha(2a) receptors. J. Biol. Chem. 1999, 274, 291082–299114.
- Briedé, J.J.; Tans, G.; Willems, G.M.; Hemker, H.C.; Lindhout, T. Regulation of Platelet Factor Va-dependent Thrombin Generation by Activated Protein C at the Surface of Collagen-adherent Platelets. J. Biol. Chem. 2001, 276, 7164–7168.
- Zhang, Y.; Liang, C. Innate recognition of microbial-derived signals in immunity and inflammation. Sci. China Life Sci. 2016, 59, 12101–12217.
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 811–818.
- Alijotas-Reig, J.; Esteve-Valverde, E.; Belizna, C.; Selva-O’Callaghan, A.; Pardos-Gea, J.; Quintana, A.; Mekinian, A.; Anunciacion-Llunell, A.; Miró-Mur, F. Immunomodulatory therapy for the management of severe COVID-19. Beyond the anti-viral therapy: A comprehensive review. Autoimmun. Rev. 2020, 19, 102569.
- Wong, J.P.; Viswanathan, S.; Wang, M.; Sun, L.Q.; Clark, G.C.; D’Elia, R. Current and future developments in the treatment of virus-induced hypercytokinemia. Future Med. Chem. 2017, 9, 169–178.
- Nunes Kochi, A.; Tagliari, A.P.; Forleo, G.B.; Fassini, G.M.; Tondo, C. Cardiac and arrhythmic complications in patients with COVID 19. J. Cardiovasc. Electrophysiol. 2020, 31, 1003–1008.
- Klok, F.A.; Kruip, M.J.H.A.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thromb. Res. 2020, 191, 148–150.
- Iba, T.; Levy, J.H.; Warkentin, T.E.; Thachil, J.; van der Poll, T.; Levi, M. Diagnosis and management of sepsis-induced coagulopathy and disseminated intravascular coagulation. J. Thromb. Haemost. 2019, 17, 1989–1994.
- Casini, A.; Fontana, P.; Glauser, F.; Robert-Ebadi, H.; Righini, M.; Blondon, M. Venous thrombotic risk related to SARS-CoV-2: Prevalence, recommendations and perspectives. Rev. Med. Suisse. 2020, 16, 9519–9554.
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 2020, 18, 1094–1099.
- Hess, D.C.; Eldahshan, W.; Rutkowski, E. COVID-19-Related Stroke. Transl. Stroke Res. 2020, 11, 3223–3225.
- Gavriilaki, E.; Brodsky, R.A. Severe COVID-19 infection and thrombotic microangiopathy: Success doesn’t come easily. Br. J. Haematol. 2020, 189, e227–e230.
- Wang, J.; Li, Q.; Yin, Y.; Zhang, Y.; Cao, Y.; Lin, X.; Huang, L.; Hoffmann, D.; Lu, M.; Qiu, Y. Excessive Neutrophils and Neutrophil Extracellular Traps in COVID-19. Front. Immunol. 2020, 11, 2063.
- Hippisley-Cox, J.; Young, D.; Coupland, C.; Channon, K.M.; Tan, P.S.; Harrison, D.A.; Rowan, K.; Aveyard, P.; Pavord, I.D.; Watkinson, P.J. Risk of severe COVID-19 disease with ACE inhibitors and angiotensin receptor blockers: Cohort study including 8.3 million people. Heart 2020, 106, 15031–15511.
- Green, C.E.; Turner, A.M. The role of the endothelium in asthma and chronic obstructive pulmonary disease (COPD). Green Turn. Respir. Res. 2017, 18, 20.
- Janga, H.; Cassidy, L.; Wang, F.; Spengler, D.; Oestern-Fitschen, S.; Krause, M.F.; Seekamp, A.; Tholey, A.; Fuchs, S. Site-specific and endothelial-mediated dysfunction of the alveolar-capillary barrier in response to lipopolysaccharides. J. Cell. Mol. Med. 2018, 22, 9829–9898.
- Tuder, R.M.; Yoshida, T. Stress Responses Affecting Homeostasis of the Alveolar Capillary Unit. Proc. Am. Thorac. Soc. 2011, 8, 485–491.
- Kaur, U.; Acharya, K.; Mondal, R.; Singh, A.; Saso, L.; Chakrabarti, S.; Chakrabarti, S.S. Should ACE2 be given a chance in COVID-19 therapeutics: A semi-systematic review of strategies enhancing ACE2. Eur. J. Pharmacol. 2020, 887, 173545.
- Varga, V.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schueqbach, R.A.; Ruschitzka, E.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 14171–14418.
- Opal, S.M.; van der Poll, T. Endothelial barrier dysfunction in septic shock. J. Intern. Med. 2015, 277, 277–293.
- Zhang, J.; Tecson, K.M.; McCullough, P.A. Endothelial dysfunction contributes to COVID-19-associated vascular inflammation and coagulopathy. Rev. Cardiovasc. Med. 2020, 21, 315–319.
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.-H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582.
- Kollias, A.; Kyriakoulis, K.G.; Dimakakos, E.; Poulakou, G.; Stergiou, G.S.; Syrigos, K. Thromboembolic risk and anticoagulant therapy in COVID-19 patients: Emerging evidence and call for action. Br. J. Haematol. 2020, 189, 846–847.
- Poterucha, T.J.; Libby, P.; Goldhaber, S.Z. More than an anticoagulant: Do heparins have direct anti-inflammatory effects? Thromb. Haemost. 2017, 117, 437–444.
- Thachil, J. The versatile heparin in COVID-19. Int. Soc. Thromb. Haemost. 2020. (In Press)
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513.
- Delabranche, X.; Helms, J.; Meziani, F. Immunohaemostasis: A new view on haemostasis during sepsis. Ann. Intensive Care 2017, 7, 117.
- Jackson, S.P.; Darbousset, R.; Schoenwaelder, S.M. Thromboinflammation: Challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood 2019, 133, 906–918.
- Huertas, A.; Montani, D.; Savale, L.; Pichon, J.; Tu, L.; Parent, F.; Guignabert, C.; Humbert, M. Endothelial cell dysfunction: A major player in SARS-CoV-2 infection (COVID-19)? Eur. Respir. J. 2020, 2001634.


