 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Atsushi Tsuji | + 1486 word(s) | 1486 | 2020-12-10 07:35:46 | | | |
| 2 | Dean Liu | -309 word(s) | 1177 | 2020-12-11 02:54:01 | | |
Video Upload Options
Tenascin-C (TNC) is an extracellular matrix glycoprotein that participates in cell adhesion, growth, migration, and differentiation. TNC is expressed at a low level in healthy adult tissues, yet it is upregulated substantially and specifically in response to tissue injury.
1. Introduction
Continuous advances in cancer therapy have led to improved survival of patients with many types of cancer [1]. Despite such advances, however, the prognosis of patients with a treatment-refractory cancer, as is often the case for pancreatic cancer, remains poor[1][2]. Most patients with refractory cancer receive multimodal therapy consisting of chemotherapy and radiation[3]. Although the outcome of patients with refractory cancer is unpredictable[2][3], anticancer treatments clearly cause damage to cancer tissues, suggesting that the cancer tissues initiate a physiological response to treatment-induced injury. Therefore, the authors hypothesized that an agent recognizing a molecule associated with that response could also mediate the delivery of anticancer drugs and radionuclides and thereby provide additional therapeutic benefit.
Tenascin-C (TNC) is an extracellular matrix glycoprotein that participates in cell adhesion, growth, migration, and differentiation[4][5][6]. TNC is expressed at a low level in healthy adult tissues, yet it is upregulated substantially and specifically in response to tissue injury[5][6]. The upregulation of TNC plays a role in tissue repair in damaged tissues but also can promote the growth, differentiation, vascularization, cell adhesion, invasion capacity, and metastatic potential of tumors[5][6]. TNC is a hexameric glycoprotein[5] that provides many potential binding sites for anticancer agents such as antibodies. Therefore, TNC is an attractive target molecule for testing our hypothesis that a drug delivery mechanism targeting a tissue injury responsive factor could increase the overall efficacy of an anticancer regimen.
Authors developed several antibodies against TNC, including three that recognize both human and murine TNC; these antibodies were named 3–6[7], 12–2–7[8], and TDEAR2[8], as shown in Figure 1. As a tumor model, a BxPC-3 pancreatic cancer xenograft tumor model was selected because BxPC-3 tumor tissues produce only small amounts of TNC in control/nontreated animals, yet substantial amounts are produced after X-ray irradiation, so this approach was appropriate to test our hypothesis. The three antibodies were radiolabeled with 111In, and changes in the uptake of the radiolabeled antibodies were evaluated in nude mice bearing tumors that had been previously subjected to X-irradiation (or were not irradiated, as a control).
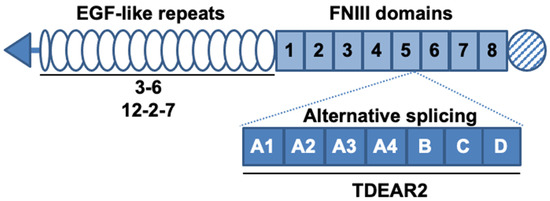
Figure 1. Schematic structure of tenascin-C (TNC) and the known binding sites of antibodies. TNC contains epidermal growth factor (EGF)-like repeats and fibronectin type III (FNIII) domains. Alternative splicing occurs between the fifth and sixth FNIII domains. The known binding sites of the three antibodies are denoted by solid lines under the domains. The antibodies 3–6 and 12–2–7 recognize the EGF-like repeats and TDEAR2 binds to the alternative splicing region.
2. X-Irradiation Increases Tumor Uptake
Authors hypothesized that an antibody recognizing a molecule associated with tissue injury repair after antitumor therapy could deliver an additional tumoricidal agent, such as a radionuclide, to cancer tissues. To test this hypothesis, the authors selected TNC as a target molecule and employed three antibodies (3–6, 12–2–7, and TDEAR2) recognizing human and murine TNC[7][8]. These antibodies were labeled with 111In, and temporal changes of the uptake of each antibody were evaluated in nude mice bearing BxPC-3 tumors exposed to X-rays, which induce TNC expression. The biodistribution studies revealed markedly increased tumor uptake of 111In-labeled antibody 3–6 with statistical significance (35% ID/g for 30 Gy vs. 15% ID/g for 0 Gy at day 1, p < 0.01). SPECT/CT imaging and autoradiographic studies provided consistent results. These findings demonstrate that an anti-TNC antibody could deliver a radionuclide to tumors, supporting our hypothesis.
Our proposed therapeutic strategy with the anti-TNC antibody coupled with an antitumor agent has three advantages. First, it targets intratumoral regions responding to damage induced by initial cancer therapy as shown in the present study, providing additional burden before successful repair. This strategy could also circumvent the problem of resistance to therapy. Second, the strategy reduces stromal barriers within the tumor microenvironment, as such barriers can inhibit the penetrance of antitumor agents, especially high-molecular-weight agents, into tumors[9]. More intratumoral stroma is formed by anticancer therapy; TNC is induced and plays a role in stroma formation[5][6][10]. Our antibody 3–6 targets upregulated TNC and could inhibit stroma formation. Third, although antibodies will generally accumulate in tumor tissues at a relatively slow rate[11], our antibody 3–6 accumulated rapidly in the tumors that had undergone therapy, indicating that radiolabeled 3–6 can deposit higher radiation doses in tumors. Taken together, the therapeutic strategy with antibody 3–6 conjugated to a tumoricidal agent, including radionuclides, has the potential to provide better outcomes when combined with conventional therapy.
Interestingly, the present study revealed a difference in tumor uptake of the three anti-TNC antibodies 3–6, 12–2–7, and TDEAR2. TDEAR2 recognizes a region in TNC derived from an alternatively spliced pre-mRNA, suggesting that the majority of TNC that is upregulated upon exposure of tumors to X-rays does not contain this region. Previous studies showed that the upregulation of TNC in response to a toxin or hapten yields the splice variants of TNC[7][12]. The splicing of TNC pre-mRNA underlies the observed spatiotemporal expression of TNC, which is associated with distinct cellular processes[13]. Our data suggest that TNC pre-mRNA is perhaps spliced in tumors after X-ray exposure, and to date, no other studies have shown this. Additional studies might provide new insights into the complexity of TNC functions during tissue repair. Although antibodies 3–6 and 12–2–7 recognize EGF-like repeats of TNC[7][8], the staining patterns for these two antibodies differed in cancer tissues, suggesting that the two recognize different epitopes. There are several glycosylation sites in the EGF-like repeats region[5], so this particular post-translational modification might affect the recognition of TNC by 3–6 and 12–2–7, leading to the different rates of uptake of the two antibodies. Further epitope analysis could reveal the reason why antibody 3–6 was taken up more aggressively by injured tumors, enabling optimization of our therapeutic strategy.
Our study has several limitations. First, the stroma of nonirradiated BxPC-3 tumors expressed only a small amount of TNC, whereas TNC is highly expressed in tumor stroma of many epithelial malignancies including pancreatic cancer[8][10]. Therefore, it will be necessary to evaluate changes in the uptake of anti-TNC antibodies in tumors that express a high level of TNC under the untreated condition. Second, upregulation of TNC expression is induced by antitumor drugs as well as by radiation[5][14]. X-rays achieve uniform distribution of radiation in cancer tissues, whereas chemotherapy and nuclear-medicine therapy result in heterogeneous distribution of drugs and radionuclides, respectively. Therefore, it will be necessary to evaluate to what extent tumor uptake changes after chemotherapy and/or radionuclide therapy to clarify what types of therapy could be combined with our proposed antibody-mediated treatment strategy.
In conclusion, the present study demonstrates that antibody 3–6 can deliver a radionuclide additionally to BxPC-3 tumors previously exposed to X-rays. This supports our concept that an antibody recognizing a specific factor, such as TNC, which is involved in tissue injury repair, could achieve the goal of delivering an additional antitumor agent to tumors during tissue repair after initial cancer therapy. A combination of antibody 3–6 with conventional cancer therapy could result in better outcomes for patients with treatment-refractory cancer.
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30.
- Gong, J.; Tuli, R.; Shinde, A.; Hendifar, A.E. Meta-analyses of treatment standards for pancreatic cancer. Mol. Clin. Oncol. 2016, 4, 315–325.
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289.
- Brellier, F.; Chiquet-Ehrismann, R. How do tenascins influence the birth and life of a malignant cell? J. Cell Mol. Med. 2012, 16, 32–40.
- Midwood, K.S.; Chiquet, M.; Tucker, R.P.; Orend, G. Tenascin-C at a glance. J. Cell Sci. 2016, 129, 4321–4327.
- Midwood, K.S.; Orend, G. The role of tenascin-C in tissue injury and tumorigenesis. J. Cell Commun. Signal 2009, 3, 287–310.
- Koyama, Y.; Norose, K.; Kusubata, M.; Irie, S.; Kusakabe, M. Differential expression of tenascin in the skin during hapten-induced dermatitis. Histochem. Cell Biol. 1996, 106, 263–273.
- Shrestha, P.; Kusakabe, M.; Mori, M. Tenascin in human neoplasia. Int. J. Oncol. 1996, 8, 741–755.
- Tarin, D. The Cancer Stroma and Its Relevance to Tumor Survival and Treatment. In Cancer Drug Delivery Systems Based on the Tumor Microenvironment; Matsumura, Y., Tarin, D., Eds.; Springer: Tokyo, Japan, 2020; pp. 3–22. ISBN 9784431568780.
- Yoshida, T.; Akatsuka, T.; Imanaka-Yoshida, K. Tenascin-C and integrins in cancer. Cell Adhes. Migr. 2015, 9, 96–104.
- Reilly, R.M. The Radiochemistry of Monoclonal Antibodies and Peptides. In Monoclonal Antibody and Peptide-Targeted Radiotherapy of Cancer; Reilly, R.M., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp. 39–100. ISBN 9780470613214.
- Matsumoto, K.; Hiraiwa, N.; Yoshiki, A.; Ohnishi, M.; Kusakabe, M. Tenascin-C expression and splice variant in habu snake venom-induced glomerulonephritis. Exp. Mol. Pathol. 2002, 72, 186–195.
- Giblin, S.P.; Midwood, K.S. Tenascin-C: Form versus function. Cell Adhes. Migr. 2015, 9, 48–82.
- Foley, K.; Muth, S.; Jaffee, E.; Zheng, L. Hedgehog signaling stimulates Tenascin C to promote invasion of pancreatic ductal adenocarcinoma cells through Annexin A2. Cell Adhes. Migr. 2017, 11, 514–523.


