1000/1000
Hot
Most Recent
 +1 point
+1 point
Ferroptosis is a type of cell death, caused by iron-mediated peroxidation of cellular lipids, that leads to tissue dysfunction in several human diseases including Friedreich's ataxia.
FRDA is an inherited autosomal recessive neurodegenerative disorder caused by the expansion of a GAA triplet-repeat sequence within the first intron of FXN gene, leading to a decrease of frataxin (FXN) [1], a mitochondrial protein involved in the synthesis of iron-sulphur clusters (Fe-S), which are essential for the activity of mitochondrial respiratory chain complexes I, II, and III, Krebs cycle enzyme aconitase and other mitochondrial enzymes [2]. Moreover, FXN is important for cellular iron homeostasis, although its role in this process has not been fully clarified yet. FRDA is characterized by early mortality due to hypertrophic cardiomyopathy. Furthermore, there is a tendency to develop type 2 diabetes in 10% of cases, due to dysfunction of pancreatic β-cells [2] and possibly to the alteration of brown adipose tissue function [3], a mitochondria-enriched tissue, which has been now ascertained to exert an anti-diabetic activity [4].
Mitochondrial energy imbalance, accumulation of mitochondrial iron, uncontrolled production of reactive oxygen species (ROS) and increased lipid peroxidation have been implicated in the pathogenesis of the disorder [5]. Such events resemble to those occurring in a new type of cell death firstly described in 2012 by Dixon et al., that is, ferroptosis [6]. Ferroptosis has peculiar and distinct characteristics compared to other forms of cell deaths. Morphologically, ferroptosis occurs with a decrease in mitochondrial mass and cristae, without the occurrence of nuclear morphological changes and DNA fragmentation [6]. Chromatin condensation, apoptotic body formation, cell shrinkage and caspase activations were not found as in the case of apoptosis; swelling of the cytoplasm and rupture of cell membrane in association with the drop in ATP levels were not found as in the case of necrosis [7]. Furthermore, formation of closed bilayer membrane structures as well as inhibition of the mTOR pathway regulating lysosomal activity were not observed as in the case of autophagy [8].
Even though the accumulation of mitochondrial iron, the increase of oxidative stress and lipid peroxidation found in several cell types with FXN deficiency (e.g., neurons from mouse models and fibroblasts from FRDA patients) [9][10][11][12] are also basically the distinctive hallmarks of ferroptosis, a possible link between FRDA and ferroptosis is emerging only recently.
In 2003 Dolma and colleagues identified erastin, a compound capable of selectively killing RAS-expressing cancer cells via a peculiar and still undescribed cell death mode [13]. Erastin blocks the Xc-system, preventing cystine cell import, thus depleting intracellular glutathione (GSH), the main non-enzymatic thiol antioxidant. Few years later, it was discovered that iron chelators were able to inhibit this type of cell death [8]. These observations led to the definition of a new iron-mediated cell death, named ferroptosis [6]. The basis of ferroptosis is the uncontrolled production of specific hydroperoxide phospholipids in the presence of free iron without the proper neutralization by the Xc-system/GSH/glutathione peroxidase 4 (GPX4) axis. The Xc-system imports cystine, the oxidized form of cysteine into cells through a 1:1 exchange with glutamate [14]. It belongs to the heterodimer amino acid transporters family and is composed of a heavy (SLC3A2) and a light chain (SLC7A11) that are finely regulated at the transcriptional level. Nuclear factor erythroid 2-related factor 2 (NRF2) positively regulates SLC7A11 [15]; by contrast, P53 downregulates its expression, leading to a decrease of intracellular cysteine and consequent increased susceptibility to ferroptosis [16]. The imported cystine is reduced to cysteine by GSH and/or thioredoxin reductase 1 and this amino acid is in turn used for the synthesis of GSH [17]. GSH can be oxidized to GSSG by GPX4, a selenium peroxidase that reduces hydroxide complexes, including phospholipid hydroperoxides and cholesterol hydroxide in the respective alcohols, thus inhibiting the lipid peroxidation chain reaction. Hence, inhibition of cystine import through erastin influences the activity of GPX4 and increases susceptibility to ferroptosis [18]. By inhibiting the lipid peroxidation chain reactions, GPX4 represents the main regulator of ferroptosis. In line with its protective role against ferroptosis-related lipid peroxidation, chemical inhibition of GPX4 (e.g., RSL3) leads to lipid peroxide accumulation and cell death [19]. Selenocysteine (SeC) is an amino acid essential for GPX4 antioxidant activity and a specific SeC-tRNA is essential for its insertion into the active site of GPX4 [20]. Studies carried out on mice that express a targeted mutation of the active SeC to Cys (GPX4_U46C) have shown that these mutated mice are viable at birth but die before weaning due to degeneration of a specific subgroup of GABA interneurons in the mouse cortex. Homozygous cells expressing GPX4_U46C are more susceptible to peroxide-induced ferroptosis with respect to controls. Selenium is therefore necessary for the correct functionality of GPX4 and for resistance to pro-ferroptotic conditions, virtually in all cells and tissues [21].
Importantly, two recent studies have identified a new mechanism inhibiting ferroptosis involving the Coenzyme Q oxidoreductase FSP1 (ferroptosis suppressor protein 1) that is independent of GSH-GPX4 system [22][23]. In particular, the suppression of ferroptosis by FSP1 is mediated by coenzyme Q10 that in the reduced form traps lipid peroxyl radicals and restrain the propagation of lipid peroxides, whereas FSP1 catalyses the NAD(P)H-dependent regeneration of coenzyme Q10.
Polyunsaturated fatty acids (PUFAs) are highly prone to a non-enzymatic lipid peroxidation that consists in a chain reaction in which the local production of free reactive oxygen-centred radicals (like HO• and HOO•) can initiate oxidation of a large portion of PUFAs, generating lipid radical (L•) species. Internal radical propagation and peroxidation of L• species finally lead to fragmentation with formation of carbonyl compounds like malondialdehyde (MDA), 4-hydroxynonenal (4-HNE), acrolein and other harmful products that can induce structural rearrangements in cell membranes [24][25][26]. Accumulation of transition metals, such as copper and/or iron oxidizes, lipids through the Fenton reaction producing an uncontrolled quantity of ROS, including HO• and HOO• that in turn increase lipid peroxidation [14]. Lipids containing PUFAs are present in the membrane of many specialized cells, such as the central nervous system (CNS) cells, and in many subcellular organelles [27]. Mitochondria are particularly rich in PUFAs whose role is to maintain the functionality of proteins and membrane transporters and to modulate the mitochondrial dynamics [28].
Acyl-CoA synthetase long-chain family member 4 (ACSL4) has been identified as an important player in lipid peroxidation. ACSL4 is one of the enzymes that activate PUFAs during phospholipid synthesis, by esterifying free fatty acids to CoA, in an ATP-dependent manner, with preference towards long-chain PUFAs such as arachidonic acid (AA). ACSL4 is also involved in the biosynthesis of phosphatidylethanolamine (PE), which contains AA. Doll et al. demonstrated that, by enriching cellular membranes with polyunsaturated fatty acids, ACSL4 enhances the sensitivity to ferroptosis [29]. As consequence, genetic or pharmacological inhibition of ACSL4 reduces the accumulation of lipid peroxides in cells, protecting against ferroptosis [30]. Since oxidized AA-PE are linked to ferroptosis, ACSL4 could be a good therapeutic target to contrast ferroptosis, especially in neurodegenerative disorders in which lipid peroxidation plays a fundamental pathogenic role [31].
Ferroptosis is also characterized by alteration of iron homeostasis, as it promotes intracellular and intra-mitochondrial iron accumulation [6]. Iron homeostasis is a finely regulated mechanism. Fe2+ released from intestinal cells or erythrocyte degradation is oxidized by ceruloplasmin to Fe3+ and bound by transferrin (TF) for serum circulation. TF-Fe3+ is recognized by the high affinity membrane protein transferrin receptor 1 (TFR1) and the complex is then internalized via clathrin-dependent endocytosis to form endosomes. The internalization of the TFR1-TF-Fe3+ complex represents a rate-limiting step of iron import under normal condition and, consistent with its function, TFR1 protein level is regulated by changes in iron status, being up-regulated or down-regulated by iron decrease or iron increase, respectively. Mechanisms regulating TFR1 expression mainly operate at mRNA post-transcriptional levels through the iron-responsive element (IREs)-iron-responsive-proteins (IRPs) system (see below for detail).
In the endosome compartment, low pH promotes transferrin to release Fe3+, which is subsequently reduced to Fe2+ by the six-transmembrane epithelial antigen of the prostate 3 (Steap3) and then transported into the cytoplasm by the divalent metal carrier 1 (DMT1). It has been recently demonstrated that DMT1 can be directly involved in mitochondrial Fe2+ influx being also localized in the mitochondrial outer membrane [32][33].
The iron transferred from endosomes into cytosol becomes then part of the labile iron pool (LIP) in the mitochondria or cytoplasm, bound to low-molecular-weight molecules [34]. Under physiological conditions, LIP is transitory and contains low iron levels that are catalytically reactive and sufficient for metabolic functions; when in excess, iron is stored by ferritin (FT). FT is a hetero-oligomeric protein complex with two different subunits: ferritin heavy chain (FTH), carrying ferroxidase activity involved in iron release, and ferritin light chain (FTL) for iron nucleation. FT is related to homeostasis of LIP by keeping under control the iron release and recycling [35].
The transport of iron out of the cell is dependent on ferroportin (FPN), which is currently the only known iron exporter. This iron transport into the extracellular compartments requires ferroxidase activity, provided by ceruloplasmin multicopper oxidase (Cp), an enzyme containing six-seven copper atoms, that oxidizes extracellular Fe2+ to Fe3+ prior the release to transferrin. Cp, as well as other multicopper iron oxidases, is essential for iron transportation, due to its molecular structure that prevents ROS formation unlike the spontaneous reaction of iron. Furthermore, multicopper oxidases have a high affinity for oxygen and this significantly reduces the rate of oxidation [36].
Homeostasis of cellular iron is under the control of two iron-regulatory proteins, namely IRP1/ACO1 and IRP2/IREB2 that are RNA-binding proteins [37]. IRPs are activated by iron deficiency and bind iron-responsive element (IRE) present in the 5′ or 3′ untranslated regions (UTRs) of numerous mRNAs encoding proteins for iron metabolism, like TFR1, DMT1, FPN, FTs subunits (FTH1 and FTL). Transcripts harbouring IREs can be modulated by IRPs in translation and/or stability, depending on IRE locations. In particular, mRNAs with IRE in the 5′ UTRs (e.g., FTs, FPN) are suppressed in translation, while transcripts with IREs in the 3′UTRs are increased in stability (e.g., TFR1, DMT1). As an example, in iron-deficient cells, activated IRPs prevent FT mRNA translation and protect TFR1 mRNA from degradation; on the contrary, under iron-sufficient conditions, low IRP activities favour FT synthesis and destabilize mRNA of TFR1, thus they properly balance iron import and store [38].
Iron amount also controls IRP functions at posttranscriptional level: IRP1 loses its RNA-binding activity and IREB2 is degraded by the proteasome system when iron is increased [38][39]. Accordingly, in mice with targeted deletion of IREB2, LaVaute and colleagues have found an increase of FT protein levels co-localizing with high iron content in degenerating neurons [40]. Therefore, it can be speculated that reducing intracellular iron concentrations could be a means to counteract the ferroptotic cascade in diseases. This is also suggested by recent experiments demonstrating that overexpression of the heat shock beta-1 protein (HSPB1) inhibits ferroptosis induced by erastin [41]. Because HSPB1 acts by decreasing endocytosis and recycling of TFR1, another strategy could be to directly silencing, the gene that codes for TFR1 [42]. Free iron is the main cause of the non-enzymatic lipid peroxidation process; for this reason, ferritinophagy, the autophagic process that leads to degradation of iron storage cell proteins, has been widely studied in relation to ferroptosis. In particular, the nuclear receptor coactivator 4 (NCOA4), which mediates the selective autophagic degradation of FT, has been recently shown to contribute to ferroptosis process by increasing iron pool and ROS through Fenton reaction [43].
Some distinctive features of ferroptosis have been recognized in many neurodegenerative diseases such as the accumulation of iron and lipid peroxide production, especially in specific regions of the central and peripheral nervous system [44]. In Alzheimer's disease (AD), the most common form of dementia, characterized by cognitive impairment and memory loss, elevated iron levels have been found in the hippocampus, which is severely impaired in patients with AD. Free iron causes ROS production through Fenton reactions, leading to massive oxidative damage in this brain region [45]. Iron chelators (e.g., Deferoxamine mesylate, DFO) were therefore used to counteract oxidative damage, reducing ferroptosis induction in AD [46]. Same strategy has been used to ameliorate the degeneration of dopaminergic neurons in substantia nigra in Parkinson's disease patients, as in this region abnormal iron accumulation is observed that is high neurotoxic [47]. Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by motor neuron dysfunction and spinal cord impairment. Ferroptotic features of dying motor neurons and increase of ferroptotic markers in blood samples have been reported, and anti-ferroptotic drugs, such as edaravone, have been proposed in parallel with other therapeutic approaches to ALS therapy [48][49].
In published literature, the topic of ferroptosis is mainly discussed in the field of cancer. Inducing iron-mediated cell death in cancer cells pharmacologically is one of the possible strategies that are going to be developed to counteract tumour aggressiveness. In this review, we have highlighted that many of the typical markers of ferroptosis are found in FRDA. Despite this, the role of ferroptosis in FRDA pathogenesis and disease progress has received relatively little attention.
The accumulation of iron inside the cell and in cellular compartments, in particular in the mitochondria, and the relative increase in oxidative stress are distinctive elements of both FRDA and ferroptosis. The production of lipid peroxides and consequent increase of their derived products, such as MDA, that have been found increased in the plasma of FRDA patients, are distinctive signs of iron-mediated cell death. The low levels of GSH found in patients and the poor activity of GPXs, in particular GPX4, suggest a strict connection between ferroptosis and FRDA. A schematic representation of the molecular factors altered in FRDA that are relevant to ferroptosis modulation is presented in Figure 1.
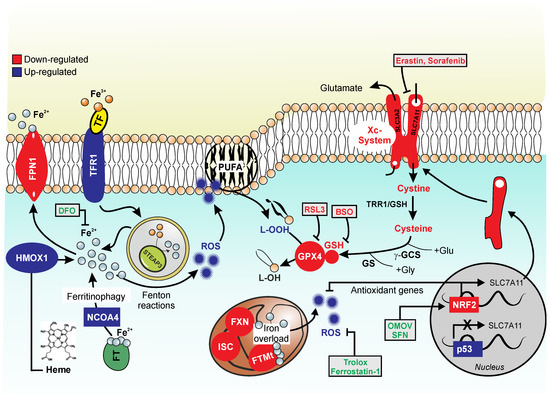
Figure 1. Ferroptotic markers in Friedreich's ataxia. The figure recapitulates the hallmarks of ferroptosis that are also characteristic of FRDA. Down-regulated factors are represented in red, while up-regulated factors are represented in blue. The figure shows reactive oxygen species (ROS) overproduction due to different causes. FXN-deficiency leads to accumulation of free iron that is not correctly bound by storage systems, such as FTMt and iron-sulfur clusters (ISC) at the mitochondrial level. Moreover, the free iron pool increases due to: augmented activity of heme oxygenase-1 (HMOX-1), which releases free iron from heme degradation; up-regulation of NCOA4, which is involved in ferritinophagy; augmented levels of transferrin receptor 1 (TFR1), which imports iron into the cell; and to insufficient iron export through ferroportin (FPN1). Free iron participates in Fenton reactions and ROS are overproduced causing oxidative stress. Upon FXN deficiency, a reduction in the Xc (SLC7A11/SLC3A2)-system-mediated cystine import also occurs, likely due to increase of P53 and decrease of NRF2 activity that represses and induces SLC7A11 transcription, respectively. This leads to the lowering of intracellular cysteine pool, thus decreasing GSH synthesis and glutathione peroxidase 4 (GPX4) activity. An overproduction of harmful lipid peroxides (LOOH) is elicited, that further contribute to ROS overproduction. Finally, the level of Nuclear factor erythroid 2-related factor 2 (NRF2), the main transcription factor involved in the expression of antioxidant genes with a strong anti-ferroptotic potential, results reduced. Ferroptosis inducers: RSL3, GPX4 inhibitor; BSO (L-buthionine sulfoximine), GSH synthesis inhibitor; erastin and sorafenib, Xc system inhibitors. Ferroptosis inhibitors: Deferoxamine (DFO), iron chelator; Trolox and Ferrostatin 1, ROS inhibitors; SFN (sulforaphane) and OMOV (omaveloxolone), NRF2 inducers.
There is no effective cure for FRDA, but many studies have proposed the use of antioxidants to counteract oxidative stress damage. Nevertheless, all the drugs currently tested have shown to have no significant benefits in restoring neural, heart and locomotor function. Hypoxia can be a viable alternative to improve oxidative status at the cellular level, even if a clinical procedure has not been still implemented. In conclusion, the molecular dynamics characterizing FRDA can be correlated to ferroptosis; therefore, therapeutic solutions that delay this type of iron-mediated cell death could provide the key to find new possible treatments for FRDA.





