1000/1000
Hot
Most Recent
 +1 point
+1 point
Immunotherapy is extensively investigated for almost all types of hematologic tumors, from preleukemic to relapse/refractory malignancies. Due to the emergence of technologies for target cell characterization, antibody design and manufacturing, as well as genome editing, immunotherapies including gene and cell therapies are becoming increasingly elaborate and diversified. Understanding the tumor immune microenvironment of the target disease is critical, as is reducing toxicity. Although there have been many successes and newly FDA-approved immunotherapies for hematologic malignancies, we have learned that insufficient efficacy due to disease relapse following treatment is one of the key obstacles for developing successful therapeutic regimens. Thus, combination therapies are also being explored. In this review, immunotherapies for each type of hematologic malignancy will be introduced, and novel targets that are under investigation will be described.
The theory of tumor immunosurveillance and the potential of immunotherapy regimens were first suggested more than 50 years ago, and, as of recently, the idea is receiving great attention due to the beneficial responses observed in certain groups of cancer patients receiving immunotherapy. Particularly, immune checkpoint inhibitors (ICIs), such as nivolumab, cause tumor remission in nonsmall cell lung cancer (NSCLC) and metastatic melanoma, whereas chimeric antigen receptor (CAR)-T cell therapy, such as tisagenlecleucel, has had great success in B cell acute lymphoblastic leukemia (B-ALL) [1]. Regardless of the success of immunotherapy in oncology, not all patients benefit from these recently developed regimens. This is mainly due to differences in the availability and immunogenicity of tumor antigens, exhaustion of cytotoxic lymphocytes, and a variety of tumor escape mechanisms. Therefore, it is important to thoroughly understand immunotherapy's mechanism of action, the tumor microenvironment, tumor immune signatures, as well as cancer cell-intrinsic genetic and epigenetic aberrations [2].
Hematologic malignancies can be driven by genetic or epigenetic changes within hematopoietic cells as well as changes in the stromal niche, including hypoxia, angiogenesis, and inflammation. Malignant hematopoietic cells can resist chemotherapy, facilitate immune evasion, and survive in the remodeled niche [3]. The goal of the current review is to summarize advances in immunotherapy approaches for the treatment of hematologic malignancies.
Cytokines, including interleukin-2 (IL-2) and IL-15, enhance the proliferation and activation of CD8+ T cells and natural killer (NK) cells [4]. In fact, interferon-alpha (IFN-α) was approved for the treatment of hairy cell leukemia, while IL-2 was introduced for the treatment of advanced melanoma in 1986 and 1998, respectively [5]. IL-2 was also infused after haploidentical NK cells were administered for treating high-risk acute myeloid leukemia [6]. An agonist of the IL-2 receptor-beta (CD122), bempegaldesleukin (NKTR-214), has been shown to increase the number and activity of CD8+ T cells without affecting Foxp3+ regulatory T (Treg) cell numbers [7]. Cytokines may also be used in combination with ICIs or CAR-T, and CAR-NK cells. For instance, methods for autologous stimulation via IL-2 have been developed for CAR constructs or through gene editing in NK-92 cells. In addition, dendritic cell (DC)-derived IL-12 plays a critical role in immune checkpoint blockade (ICB) therapy [8]. Overall, cytokines boost immune system responses, including antitumor immunity.
T cells employ two mechanisms for the killing of target tumor cells: one is through antigen-specific signaling via T cell receptors (TCRs), and the other is through antigen-nonspecific signals. The latter is associated with costimulatory receptors (e.g., CD28) or coinhibitory receptors (e.g., CTLA-4 and PD-1). Cytotoxic T cells and NK cells are suppressed upon the engagement of coinhibitory receptors, allowing for the immune escape of target tumor cells [9]. Thus, blocking inhibitory checkpoints with ICIs can harness immune cells to effectively attack tumor cells. It has been demonstrated that ICIs can indeed successfully extend survival for months or years in patients who would otherwise survive for less than a year on other recommended treatment regimens [10]. Since 2011, seven ICB therapeutics, including ipilimumab, nivolumab, and pembrolizumab, have received FDA approval for the treatment of metastatic melanoma, advanced NSCLC, Hodgkin lymphoma, and other malignancies. [11].
CTLA-4 is expressed on activated T cells, including Treg cells. Upon T cell receptor (TCR) activation, CTLA-4 is upregulated and interacts with CD80 on antigen-presenting cells (APCs), resulting in T cell-intrinsic suppression [12]. For CD80 binding, CTLA-4 competes with T cell costimulatory receptor CD28, indicative of the regulatory role of CTLA-4 in T cell activation [13]. In addition, CTLA-4 engagement in Treg cells can induce the release of an immunosuppressive mediator indoleamine 2,3-dioxygenase (IDO), suggesting that CTLA-4 plays a crucial role in Treg-mediated immune tolerance [14][15]. Recently, CTLA-4 recycling is considered to be an important factor for preventing immune-related adverse events (irAEs) during the use of anti-CTLA-4 monoclonal antibodies (mAbs) in cancer. CTLA-4 is colocalized with lipopolysaccharide-responsive and beige-like anchor (LRBA) protein in endosomes, where it can be recycled or directed to the lysosome for degradation [16]. Interestingly, the irAE-prone anti-CTLA-4 mAbs (e.g., ipilimumab) directed lysosomal degradation of surface CTLA-4 by preventing its interaction with LRBA. Recently, a novel anti-CTLA-4 mAb has been developed. It was engineered to dissociate from CTLA-4 in endosomal vesicles due to its pH level, leading to the improvement of irAE upon using the anti-CTLA-4 mAb. It demonstrates that the targeting of CTLA-4 needs to be tightly controlled [17].
PD-1, upon engagement of its ligands PD-L1 or PD-L2, recruits tyrosine phosphatase SHP2 and inhibits the T-cell-receptor-mediated intracellular activation signaling cascade [18]. CD28 is known to be the most sensitive target of PD-1-SHP2. PD-1 also represses the expression of genes transcribed following strong TCR signals, such as those encoding cytokines and effector molecules [19]. The PD-L1/CD80 cis-heterodimer can be found on APCs, affecting the trans-interaction with PD-1, CTLA-4, or CD28, suggestive of a competitive interaction. Detailed investigations on the use of ICIs in hematologic malignancies will be further discussed in the next section.
In general, chemotherapy employs a cytotoxic compound that affects the whole body. Thus, targeted drug delivery systems have long been desired and optimized to improve the efficacy of chemotherapy agents by minimizing adverse effects in healthy tissue. The introduction of monoclonal antibodies allowed researchers to employ their specificity as a mechanism for targeted drug delivery. Hence, antibody–drug conjugates (ADCs) were developed by chemical conjugation of a cytotoxic agent to a tumor-targeting antibody [20]. The antibody recognizes tumor cell-specific or cell-enriched antigens and delivers a highly potent DNA-binding agent, attached to the antibody via a cleavable linker [21].
In 2000, the first ADC, gemtuzumab ozogamicin, was approved for treating acute myeloid leukemia [22]. It comprises an anti-CD33 antibody conjugated to calicheamicin, a potent DNA-targeting agent. It was recently reapproved by the FDA, after being withdrawn in 2010. Tagraxofusp, a fusion protein consisting of IL-3 and diphtheria toxin, was FDA-approved for blastic plasmacytoid dendritic cell neoplasm (BPDCN) in 2018. BPDCN is a rare and clinically challenging hematologic malignancy; however, a breakthrough has been made by detecting CD123, a surface receptor for IL-3, which was overexpressed in the tumor cell. The overall response rate was 90% with tagraxofusp and further studies to improve outcomes will be continued [23].
ADCs have also been successful in solid tumors; for example, ado-trastuzumab emtansine for the treatment of HER2-positive breast cancer [24]. More clinical trials are underway for ADCs against solid tumors. It has been suggested that the level of target antigen expression in tumor cells and the selection of patients based on diagnostic test results are essential for ADC treatment efficacy.
Bispecific antibodies may recognize both T cells or NK cells and tumor-associated antigens (TAA), thus directing immune cells to target cancer cells more effectively. Typically, bispecific T cell engagers (BiTEs) have been developed against CD33/CD3 and CD123/CD3 for the treatment of hematologic malignancies [25][26]. Similarly, bi- and trispecific NK cell engagers (BiKE and TriKE) MLLhave also been developed, linking activating NK receptors (e.g., CD16) to TAAs. The promising results with this strategy led to the achievement of the FDA approval of blinatumomab for treating B-ALL [27]. Blinatumomab is a BiTE consisting of variable regions of anti-CD3 and anti-CD19 and has brought a significant treatment advancement for patients with relapsed or refractory and/or minimal residual disease-positive B cell ALL [28][29]. Nonetheless, an obstacle has been suggested for employing BiTEs where the therapeutic efficacy could be impeded due to T cell exhaustion or anergy [30]. TAA-positive tumor cells often upregulate the surface level of PD-L1, and the activity of T cells recruited to PD-L1-high tumor cells would be compromised via the PD-1 signaling cascade. Thus, combination therapy with PD-L1/PD-1 blockade and BiTE antibodies has been investigated, and it has resulted in enhanced T cell activity, as indicated by higher IFN-gamma production [31].
CAR-T cell therapy is an adoptive cellular immunotherapy using genetically modified lymphocytes. Prior to infusion into the patient, T cells are collected from the patient, expanded in a bioreactor, and modified to express a specific CAR [32]. The CAR consists of an extracellular domain, a single-chain variable fragment (scFv) that recognizes the tumor antigen, a transmembrane domain, and an intracellular T cell activation domain, most often CD3z. In addition to CD3z, intracellular domains of T cell receptor costimulatory molecules, such as CD28 and/or 4-1BB, are necessary to ensure the persistence and efficacy of CAR-T cells, and the incorporation of these domains led to the development of second- and third-generation CARs [33][34]. Subsequently, fourth-generation CARs called T cells redirected for antigen-unrestricted cytokine-initiated killing (TRUCKs) have also been developed. These can further induce cytokine production or apoptotic protein expression upon activation [35].
The FDA approved three CAR-T cell therapies in 2017 and 2020, namely tisagenlecleucel, axicabtagene ciloleucel, and brexucabtagene autoleucel [36][37]. They express an scFv derived from the mouse monoclonal antibody FMC63, which specifically recognizes human CD19. With regard to the costimulatory intracellular domain, axicabtagene ciloleucel and brexucabtagene autoleucel are composed of CD28, whereas tisagenlecleucel contains 4-1BB. Although the scFv is a critical factor that determines the target of CAR-T cell therapy, the effect of other domains, including the linker [38], hinge [39], transmembrane, and intracellular/costimulatory domains [40][41], should be thoroughly investigated, as it could modulate efficacy and associated adverse events.
Although CAR-T cell therapies are innovative and have been successful in certain patients, about 70% of patients who receive CAR-T cells fail to respond or relapse after therapy [42][43]. Therefore, extensive efforts have been made to enhance CAR-T cell activity through the development of next-generation CAR constructs and combination therapy with ICB or other CARs. For instance, various CAR-T cells have been introduced that can generate immunostimulatory ligands and cytokines, such as the CD40 ligand [44], Fms-related tyrosine kinase 3 (FLT3) ligand [45], IL-12 [46], and IL-18 [47], upon engagement. Immune checkpoint receptor PD-1 can also be upregulated in activated CAR-T cells, and, recently, various studies have reported that suppression of PD-1 in parallel to CAR-T therapy could be beneficial. Further, genome engineering techniques such as CRISPR/Cas9 [48] and TALEN [49] were used to generate PD-1-deficient CAR-T cells. CAR-T cells were also modified to secrete PD-1-blocking scFv, which improved the antitumor response. A dual CAR-T targeting both CD19 and CD20 by transduction of CAR containing tandem scFv to T cells has also been investigated, which results in an elicited antitumor response [50]. In addition, the limited autologous T cell expansion could be overcome through the development of TCR alpha removed CAR-T cells or allogeneic CAR-NK cells, which are being investigated [51].
CARs can be introduced into NK cells, generating CAR-NK cells, which are generally thought to be safer than CAR-T cells [52]. Recently, promising results from a clinical trial using anti-CD19 CAR-NK cells were reported [53]. CAR-NK cells were infused to patients with relapsed or refractory CD19-positive hematologic malignancies, and complete remission (CR) was seen in 64%. The CAR construct possesses IL-15, and the authors suggested that this might have played an important role in the persistence of CAR-NK cells with functional activity in vivo for several months. Notably, the NK cells were derived from HLA-mismatched cord blood, suggesting the advantages of employing NK cells over T cells. More importantly, the recipients did not represent significant adverse events by CAR-NK cells, such as cytokine release syndrome.
Hematologic malignancies develop as a result of genetic and epigenetic changes that accumulate in hematopoietic cells. There are several different types of hematologic malignancies with different etiologies. It can be categorized by the affected hematopoietic cell type as well as the place where the tumor occurs. In this section, the pathophysiology of hematologic malignancies and the relevant immunotherapies will be summarized.
Developing effective immunotherapy for AML has been challenging. AML cells are heterogeneous, which contributes to the inability of the immune system to recognize tumor-specific markers [54][55]. Indeed, AML cells can develop immune escape mechanisms effectively avoiding death, for instance, by suppressing NK cells or reducing the expression of certain surface receptors. AML cells also increase expression of inhibitory immune checkpoints including PD-L1, PD-L2 [56], CD47 [57], and CD70 [58]. Therefore, many trials on immunotherapies and/or combination therapies are being conducted with promising results. Anti-PD1 or anti-CTLA4 therapy following disease remission after chemotherapy has been tested for eliminating measurable residual disease (MRD), demonstrating strong T cell responses against AML [59]. The interaction between CD70 and CD27 is one of the immune escape mechanisms along with the increased frequency of Treg cells or enhanced clonal expansion of AML cells via TRAF2- and TNIK-mediated canonical Wnt pathway activation [60]. A CD27-targeting mAb (varlilumab) effectively eliminated CD27-expressing lymphoma and leukemia [61]. An anti-CD70 mAb (cusatuzumab) has also been developed and proved to be a promising therapeutic approach in preclinical models of AML [62].
Since the first approval of ADC, gemtuzumab ozogamicin, comprising an anti-CD33 antibody conjugated to calicheamicin [22], approximately 80 ADCs have been developed and assessed in nearly 600 clinical trials [63]. The first ADC was approved for AML and vadastuximab talirine, also known as SGN-CD33A, in combination with hypomethylating agents or pyrrolobenzodiazepine, was evaluated in clinical studies for newly diagnosed or relapsed AML as well as for newly diagnosed MDS. SGN-CD123A is also in clinical development for treating AML [64].
A BiTE, anti-FLT3/CD3 (AMG-427) is currently being evaluated in a clinical study (NCT03541369, phase I) [65]. Anti-CD33/CD3 (AMG330) [66] and anti-CD123/CD3 [67] have also been suggested as treatments for AML. FLT3 mutation is the most common mutation observed in AML (about 30%) and induces the ligand-independent, constitutive activation of the receptor tyrosine kinase, enhancing cell survival. Similarly to FLT3 inhibitors (e.g., gilteritinib), antibodies against FLT3 are also effective for reducing cell growth. Furthermore, FLT-3-targeting BiTEs are able to recruit cytotoxic T cells to destroy tumor cells. CD123 is an IL-3 receptor subunit and is considered an LSC marker. CD123 is expressed on CD34+CD38- AML cells, and these cells with CD34+CD38-CD123+ were able to engraft in immunodeficient mice [67]. Thus, CD123 represents a promising target molecule for the detection of AML cells without affecting the healthy bone marrow cells. The overexpression of CD123 is associated with the constitutive phosphorylation of STAT5, accelerated cell proliferation, and reduced apoptosis [68]. Other antibodies against C-type lectin domain family 12 member A (CLL-1) [69], mainly expressed in AML LSC specifically, CD47 [70], and IL-1 receptor accessory protein (IL1RAP), which stimulates oncogenic activity in AML through activation of the innate immune signaling pathway [71], have also been suggested as therapeutic candidates.
The antigens described above can be exploited as targets of CAR-T cells. CD33 CAR-T therapy was tested in combination with autologous CD33-knockout bone marrow transplantation using a gene-editing tool, such as CRISPR/Cas9 [72]. Since CD33 is expressed in normal HSCs but has no relevant function, this approach was feasible. CD123 CAR-T cells are also in clinical development. CD117 CAR-T cells were recently reported to efficiently eliminate AML blasts as well as CD117+ healthy HSCs in the AML model [73]. CD117, a receptor tyrosine kinase to which stem cell factor binds, is expressed on hematopoietic precursors; however, it may remain overexpressed following malignant transformation in HSCs [74][75]. In a recent study, the CAR was modified to comprise an anti-CLL-1 scFv linked to an anti-CD33 scFv via a self-cleaving P2A peptide, resulting in the expression of both functional CARs on the T cell surface [76]. Another dual CAR-T cell therapy employing anti-CD123/CLL-1 is currently being evaluated in a clinical trial (NCT03631576). Similarly to CAR-T cell therapy, adoptive cell transfer is a promising treatment method for the stimulation of patients' immunity. In addition, harnessing NK cells for adoptive cell transfer is feasible, as alloreactive NK cells from the donor can suppress leukemic cells and LSCs, as shown in a patient-derived xenograft (PDX) animal model with PARP inhibitor cotreatment [77]. Moreover, personalized dendritic cell vaccines, namely DC/AML fusion cells, can be infused into patients, resulting in AML remission [78]. Vaccination and/or transfer of antigen-specific CD8+ T cells with WT1 extended overall survival in AML [79][80]. Taken together, with the technical advances in gene editing and ex vivo expansion of human immune cells, adoptive immune cell transfer methods are quickly improving and contributing to personalized medicine.
Among disease types of MPN, the Philadelphia chromosome-negative MPNs, PV, ET, and PMF are considered chronic and inflammation-related diseases, which implicates the dysregulation of the immune system. Thus, immunotherapies are tested and conducted to treat these types of MPN [81]. IFN-alpha has been used for the management of myelofibrosis in MPNs for over 30 years. It has recently been recommended as an alternative to ruxolitinib for the treatment of low-risk MPNs by the National Comprehensive Cancer Network [82]. IFN-alpha has antiproliferative, proapoptotic, and immunoregulatory effects, which could modulate disease symptoms related to the aberrant immature megakaryopoiesis and granulocytosis observed in MPNs [83]. In line with this notion, a long-acting novel IFN-alpha, ropeginterferon-alpha 2b, has been approved for treating PV patients in Europe.
Interestingly, identifying neoantigens in MPNs for monoclonal antibody therapies or utilizing ICIs may lead to better results in MPNs in comparison to MDS and AML. It has been reported that JAK2 mutations can trigger the upregulation of PD-L1 on myeloid cells and PD-1 ICB improved disease symptoms in a human MPN xenograft murine model [84]. Notably, neoantigens can be also identified in association with mutations of JAK2V617F (>50%), MPL (3–5%), and CALR (20–30% in ET and PMF) in MPN, which would elicit tumor-specific T cell responses [85][86]. However, treatment with IFN-alpha or ICB has limited effects on patients who are refractory to JAK2 inhibitors, suggesting that there is an unmet need for the treatment of MPN through immunotherapies [87].
The above-described features of HL have led to the investigation of the efficacy of PD-1 ICIs in HL. In a clinical trial, the rate of progression-free survival at 24 weeks was 86%. Nivolumab substantially improved disease symptoms in patients with previously heavily treated relapsed or refractory HL and received FDA approval in 2016, followed by the approval of pembrolizumab in 2017 [88]. It is thought that compared to other hematologic malignancies, the benefit of PD-1 blockade is most clearly observed in HL patients.
RS cells are multinucleated large cells found in the lymph nodes of individuals with HL and used as a diagnostic measurement. Thus, RS cells have been thought to be a therapeutic target for treating HL. They are known as having B lymphocyte origin; however, the RS cells of classical HL fail to express most B cell functional genes and markers, such as CD20. RS cells are CD30-positive, and therefore, antibodies against CD30 are considered a therapeutic regimen. Brentuximab vedotin, an ADC that consists of an antibody against CD30 and a potent microtubule-disrupting agent, namely auristatins, was approved for treating HL and anaplastic large-cell lymphoma (ALCL) in 2011. Recently, trials on the combination of nivolumab and brentuximab vedotin in patients with relapsed or refractory classical HL showed a significant increase in complete remission rate compared with treatment with either nivolumab or brentuximab vedotin alone [89]. Brentuximab vedotin induced the depletion of CD30-positive RS cells, and the administration of nivolumab resulted in an increase of the T cell subset in peripheral blood [90].
In contrast to HL, PD-1 blockade has not resulted in obvious clinical responses in patients with NHL, including DLBCL and FL. Combination therapy with nivolumab and ibrutinib (a Bruton's tyrosine kinase inhibitor), has also been assessed in relapsed or refractory DLBCL and FL, as well as CLL. However, the overall response was not different from that of ibrutinib monotherapy [91]. The tumor microenvironment of DLBCL has been described as immunologically "cold" based on transcriptional and histological studies, indicating low immunogenicity [92]. Of note, the tumor immune microenvironment is strongly associated with ICB efficacy, and there are some NHL subtypes exhibiting good response rates to PD-1 blockade. For instance, it was recently reported that patients with EBV-positive NHL responded to pembrolizumab more efficiently than those with EBV-negative NHL, presumably due to high PD-L1 expression in EBV-positive NHL [93]. Up to 25% of DLBCL patients harbor PD-L1 gene alterations, resulting in PD-L1 overexpression. These cases represent low progression-free survival following front-line chemotherapy despite the high infiltration of clonal T cells. Thus, PD-1 blockade has resulted in good responses in these patients [94]. Importantly, a phase II study on the treatment of FL patients with the front-line use of a combination of nivolumab and rituximab resulted in an objective response rate (ORR) of 84% and CR of 47%, suggesting that the patients' disease status might be important with respect to ICB response [95].
Exceptional successes of CD19 CAR-T cell therapies in clinical trials have led to achieving their FDA approvals. The first FDA-approved CAR-T, tisagenlecleucel, was treated for B cell ALL patients at 25 years of age or younger. Encouraging results have been reported with an ORR of 81% and relapse-free survival of 66% at 18 months [42]. In the following year, 2018, the same drug was approved for treating adults with relapsed or refractory DLBCL. Axicabtagene ciloleucel, another CD19 CAR-T cell, was also approved for relapsed or refractory large B cell lymphoma and DLBCL in 2017. A long-term follow-up of a phase I/II clinical trial using axicabtagene ciloleucel reported an ORR of 83% and a CR of 40% in RR DLBCL with the response persisting for over two years [96]. In 2020, brexucabtagene autoleucel was approved for treating mantle cell lymphoma. It consists of a similar CAR construct to axicabtagene ciloleucel, but the final CAR-T cell product has been improved by using a different autologous T cell enrichment process. It showed an ORR of 87% and a CR of 62% at six months [37].
Despite the overall long-term survival of up to 30% of treated patients, the remaining patients experience adverse events or relapse after CAR-T cell therapy. The major mechanisms for relapse are considered to be the poor persistence of CAR-T cells and loss of CD19 antigen expression on the malignant cells. The technical and biological obstacles for the production of autologous CAR-T cells also have to be overcome. Various novel strategies, including fourth-generation CAR-T cells, inducible CAR-T cells, and/or multiple antigen-targeting CAR-T cells, have been described for overcoming limited efficacy [97].
Although an early clinical trial of combination therapy with pembrolizumab, lenalidomide, and low-dose dexamethasone reported good efficacy (ORR 44%) in relapsed and refractory MM patients [98], the following phase III trials revealed an unfavorable risk profile [99][100]. Recently, single-cell RNA-seq analysis of the MM tumor microenvironment indicated that a senescent and dysfunctional T cell subset is enriched in the bone marrow [101]. MM predominantly grows in the bone marrow, and the unique tumor microenvironment might prohibit reinvigoration of T cells in response to PD-1 blockade [102]. Furthermore, MM progression can trigger an increase in immunosuppressive subsets, such as myeloid-derived suppressor cells (MDSCs) and Treg cells. This led to a clinical trial of daratumumab (anti-CD38 mAb, against CD38+ Treg cells) in combination with an anti-PD-1 mAb for relapsed and refractory MM, which was terminated due to increased adverse events and less benefit [103].
In 2020, belantamab mafodotin (Blenrep) was approved as an ADC for the treatment of patients with relapsed or refractory MM who have received at least four prior therapies, including an anti-CD38 monoclonal antibody, a proteasome inhibitor, and an immunomodulatory agent. The drug is a monoclonal antibody specific to B cell maturation antigen (BCMA) and linked to toxic drug auristatin F. This ADC attracted attention as it was the first approved immunotherapy targeting BCMA. BCMA is an important target for treating MM, and BiTEs targeting it are also being developed.
Several CAR-T cell therapies are being investigated for MM, and the most advanced one targets BCMA. BCMA CAR-T therapy had outstanding results in heavily pretreated MM patients [104], and it was filed for US FDA approval in March 2020. The overall response rate of BCMA CAR-T cell therapy was 73.4% with 31.3% of patients achieving a complete response. SLAMF7 is a member of the signaling lymphocytic activation family of receptors and regulates the immune system. It is enriched on malignant plasma cells' surface, while also expressed on other immune cells, including NK cells, T cells, B cells, and macrophages, but not on HSCs or nonhematopoietic cells [105]. Notably, an anti-SLAMF7 mAb (elotuzumab) in combination with lenalidomide exhibited a response without significant adverse events in MM, leading to the development of SLAMF7 CAR-T cells [106]. Additionally, CD44v6, an isoform of the hyaluronate receptor, is also targeted by CAR-T therapy for treating patients with MM. Remarkable antimyeloma effects were reported for CD44v6 CAR-T cells in a mouse model [107][108].
A summary of types of hematologic malignancies, incidence rates, and immunotherapies that received FDA approval is shown in Figure 1.
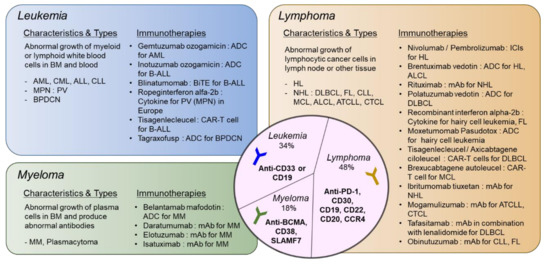
Figure 1. Types of hematologic malignancies and immunotherapies that received FDA approval. Hematologic malignancies are categorized into leukemia, lymphoma, and myeloma. Disease characteristics and subtypes are described and currently available immunotherapies approved by FDA are listed. In the middle circle, targets for immunotherapeutic approaches specific to each disease type are shown. ADC: antibody–drug Conjugate; ALCL: anaplastic large-cell lymphoma; ALL: acute lymphoblastic leukemia; AML: acute myeloid leukemia; ATCLL: acute T cell leukemia/lymphoma; B-ALL: B cell acute lymphoblastic leukemia; BPDCN: blastic plasmacytoid dendritic cell neoplasm; CAR: chimeric antigen receptor; CLL: chronic lymphocytic leukemia; CML: chronic myeloid leukemia; CTCL: cutaneous T cell lymphoma; DLBCL: diffuse large B cell lymphoma; FL: follicular lymphoma; HL: hodgkin lymphoma; ICI: immune checkpoint inhibitor; MCL: mantle cell lymphoma; MM: multiple myeloma; MPN: myeloproliferative neoplasm; NHL: non-Hodgkin lymphoma; PV: polycythemia vera. please refer to the manuscript for other abbreviations. The list of drugs can be searched at https://www.cancer.gov/about-cancer/treatment/drugs/.





