 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Michael Webb | + 6491 word(s) | 6491 | 2020-10-19 11:16:43 | | | |
| 2 | Vicky Zhou | -2770 word(s) | 3721 | 2020-11-02 09:27:40 | | | | |
| 3 | Vicky Zhou | -2770 word(s) | 3721 | 2020-11-02 09:52:23 | | |
Video Upload Options
Declining mitochondrial function, reflected in defects in ATP synthesis and increased generation of toxic reactive oxygen species is a universal feature of natural ageing. It accompanies the other hallmarks of ageing which include progressive loss of function in multiple organs, sarcopenia and increasing maladaptive low-grade inflammation. These end in death, which is a cumulative result of loss of function, leading to either increased vulnerability to environmental hazards such as predation and disease or to failure of critical organ systems such as the heart, liver or kidney. Several processes that may contribute mechanistically to age related degeneration have been identified, including oxidative damage, accumulation of toxic protein aggregates, autoinflammatory processes, loss of stem cell populations and an increasing load of malfunctional senescent cells. Mitochondrial dysfunction has connections with each of these processes. The following is a brief overview of some of these connections.
1. Mitochondrial DNA and Biogenesis
Of the approximately 1500 proteins constituting mitochondria, only a small number are encoded in mitochondrial DNA and translated on mitochondrial ribosomes. The mammalian mitochondrial genome encodes a set of 22 mitochondrial specific tRNAs, 2 ribosomal RNAs and 13 polypeptides, all of which are components of the electron transport chain [1][2]. It is embodied in a circular DNA molecule of 16.5 kb diameter, which is present in copy numbers between 100 and 10,000 per cell, depending on the cell type. Mitochondrial biogenesis requires a close co-ordination of the synthesis, import and localization of the nuclearly encoded cytoplasmically synthesized mitochondrial proteome with the mitochondrial endogenous protein synthesis system.
Global mitochondrial proteomic changes are observed during ageing [3][4]. Down regulation of genes associated with electron transport activity with age is one common theme [5] and the result of reduced expression of newly synthesized mitochondrial components may be exacerbated by an increasing inability of the ageing cell to rid itself of old and damaged mitochondria [6]. Of the 13 mitochondrial translation products examined by Short et al. [3] in normally ageing human, nine were significantly lower in older subjects, the declines in COX3 and COX4 being roughly of the same order of magnitude as the decline in ATP synthetic capacity. They also showed that in normally ageing human, mtDNA and mtRNA abundance declined together with mitochondrial ATP production.
Mutations in mitochondrial DNA, (whether in genes encoding subunits of the electron transfer chain or in mt tRNA or rRNA) are frequently associated with a decreased efficiency in ATP synthesis and increased generation of ROS, reflecting the specialized contributions of mitochondrially encoded components to the generation of ATP via electron transport. As these same processes accompany ageing, it has usually been assumed that mitochondrial mutations contribute to the ageing process.
Mutation rates of mitochondrial DNA are estimated to be approximately 10–70-fold higher than nuclear DNA [7][8]. mtDNA lies near the sites of reactive free radical generation, exists in more vulnerable single stranded form for much of the replication cycle and unlike nDNA, is not highly compacted into more protected chromatin. However, the emerging consensus is that in spite of this, the major source of mutation in mtDNA is copying errors introduced during replication [8][9].
Synergistic interactions between different mtDNA mutations within the same mitochondrion may affect ageing and function even when the individual mutations have no measurable effect on fitness. Reichart et al. [10] studied the effects of mtDNA mutations in a complex IV component and in the mttRNAArg gene on lifespan, learning and memory. Single point mutations in either gene had little impact on these parameters but mice with the combination of both mutations had a reduced life span and showed deficits in learning and memory.
The expression of nuclear genes contributing to mitochondrial biogenesis requires transcriptional activators and co-activators. The former can bind recognition sites in the promoters of target genes and initiate transcription. The major transcriptional activators of mitochondrial biogenesis programs so far identified are Nrf-1 and Nrf-2 (also known as GA binding protein, GABP), Estrogen-related-receptors (ERR—α,β and γ), CREB, FOXO, PPARδ and cMyc [11][12][13] and their regulation by physiological stimuli is indicated in Figure 1. Their targets may include both mtDNA and nDNA encoded genes.
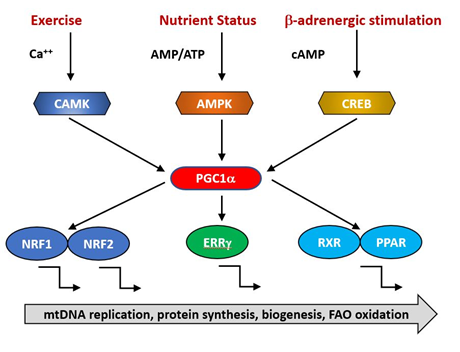
Figure 1. PGC1α is a transcriptional co-factor that regulates the transcription of nuclearly encoded mitochondrial proteins by interacting with transcription factors such as Nrf1/2, ERRg and the PPAR family. It acts as a hub to integrate signals from several pathways that monitor the energetic and nutritional status of the cell, including AMP-activated protein kinase (AMPK), calcium/calmodulin-dependent protein kinase (CAMK) and cAMP-response element binding protein (CREB).
The activities of these transcription factors are coordinated by transcriptional co-activators, of which the best studied is PGC1α, together with its related coregulators PGC-1β and PRC. Positive and negative regulators of mitochondrial protein synthesis that are responsive to such physiological factors as exercise, hormonal status and nutritional status converge on PGC1α, commonly referred to as the master regulator of mitochondrial biogenesis. It is subject to post-translational activation by a wide array of upstream elements including 5′ AMP-activated protein kinase (AMPK), glycogen synthase kinase-3 (GSK-3) and Sirtuin1 (SIRT1). Collectively, these transduce signals related to the physiological status and energy demand of the organism. Reduced AMPK activity has been reported in aged animals [14] and is directly linked to age-related insulin resistance and impaired fatty-acid oxidation [15][16][17] . This makes the point that an overall reduction in mitochondrial function need not imply a specific primary defect in mitochondria but rather a failure in the integration and transmission of the upstream physiological monitoring systems.
2. Biochemistry of Mitochondrial Ageing
Changes in levels of several key metabolites are seen with age. These include Coenzyme Q (2,3 dimethoxy-5-methyl-6-decaprenyl-1,4-benzoquinone; CoQ10, ubiquinol), NAD and some components of the Krebs Cycle. CoQ a small lipophilic molecule with key roles in electron transport and anti-oxidant activity. Levels of Coenzyme Q decline with age in some but not all tissues in both rodents [18] and human [19].
The Krebs cycle has been proposed as an important component of anti-oxidant defenses via the formation of 2-oxoglutarate (also known as α-ketoglurate) [20]. In addition to its anti-oxidant roles, 2-oxoglutarate is also a substrate for 2-oxoglutarate-dependent dioxygenases (2-OGDO) [21]. These constitute a family of enzymes regulating DNA and histone methylation, including Ten-Eleven Translocation (TETs) and Jumonji C domain containing (JmjC) demethylases and they are inhibited by two other Krebs cycle intermediates, succinate and fumarate. The regulation of enzymes controlling epigenetic changes in DNA provides a potential link between age related changes in mitochondrial Krebs cycle activity with changes in nuclear gene expression that may contribute to the ageing process [22].
Krebs cycle intermediates may play additional roles in signaling mechanisms relevant to some disorders of ageing. Thus, succinate is a ligand for a G protein coupled receptor, GPR91 and a related receptor GPR99 recognizes oxoglutarate [23]. Succinate increases blood pressure, an effect not seen in GPR91 knockout mice and there is therefore a plausible link between alterations in Krebs cycle intermediates and some disorders associated with ageing such as renal hypertension and atherosclerosis [24]. GPR91 is also expressed in ganglion cells of the retina. During the development of diabetic retinopathy, succinate levels increase in the retina, activating GPR91 and up-regulating VEGF signaling [25] which plays a central role in mediating microvascular and macrovascular pathology in diabetes.
One striking biochemical correlate of ageing with a clear connection to mitochondrial function is the decrease with age of nicotinamide adenine dinucleotide (NAD); in vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences [26]. The direct link between NAD levels and ageing is indicated by the observation that NAD repletion enhances life span in mice [27]. NAD decreases in natural ageing are linked to reduction in expression of some NAD producing enzymes (such as the enzyme nicotinamide phosphor-ribosyltransferase (NAMPT)), and increases in NAD consuming enzymes such as PARP and CD38 (cluster of differentiation 38) as indicated in Fig 2. Under acute stress conditions, PARP inhibition may preserve NAD and ATP levels and allow cellular survival [28]. However, PARP activity chronically correlates with species-specific lifespan [29] and centenarians have higher PARP activity than a control group of subjects between the ages of 20 and 70 [30]. CD38 is a major consumer of NAD [31], is induced under inflammatory conditions [32] and its expression increases with age [33][34]. As general systemic inflammation also increases with age [35][36][37][38][39] a causal relationship may exist between increased CD38 expression and decreasing NAD levels. The major age-related changes in NAD producing and consuming enzymes are indicated in Figure 6. The central role of CD38 is also supported by the observation that a potent CD38 inhibitor ameliorates metabolic dysfunction in aged mice [40].
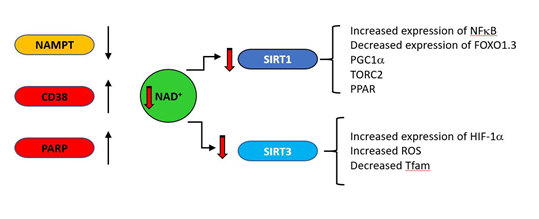
Figure 2. During ageing, upregulation of NAD consuming enzymes such as CD38 and poly (ADP-ribose) polymerase (PARP) and down regulation of the synthetic enzyme nicotinamide phosphor-ribosyltransferase (NAMPT) result in reduced levels of NAD. This is results in lower activity of the NAD–dependent deacylases (sirtuins). SIRT1 and SIRT3 regulate a variety of mitochondrially related functions whose age related change is regarded as deleterious, including decreases in transcription of genes such as PGC1a, PPAR, TFAM and FOXO1.3 and upregulation of NFκB.
Cardiolipin is a non-bilayer forming phospholipid dimer with a wide diversity of molecular forms. It is expressed almost exclusively in mitochondria, where the vast majority is found on the inner mitochondrial membrane [41]. Its structure confers unique biophysical properties on the molecule [204] and it is a key component of multiple mitochondrial activities. Its intimate association with the inner membrane sites of ROS generation renders cardiolipin particularly susceptible to oxidative damage in the form of lipid peroxidation [42]. Oxidation of cardiolipin impairs its function in bioenergetic processes [43][44]. Under stress conditions, cardiolipin may redistribute from the inner mitochondrial membrane to the outer membrane, where its exposure can trigger either mitophagy [45] or apoptosis [46][47]. Decreases in cardiolipin content with age have been reported in mitochondria of brain, liver and heart [48][49][50] and this is likely a contributor to age related decreases in energetic efficiency.
3. Oxidative Damage Theory
An early hypothesis to explain cellular and organismal impairment with age linked the occurrence of mutations in mitochondrial DNA with the production of toxic free radicals. As indicated in Figure 3, mitochondria are the major source of free radical production in most cells, as a result of electron leak from complexes I and III of the electron transport chain, these electrons combining with molecular oxygen to produce the unstable reactive superoxide radical (.O−). This can directly attack molecules such a protein, lipids and nucleic acids and can give rise to various additional reactive species such as hydroxyl (•OH) and peroxynitrite ONOO−, not itself a free radical but a potent oxidizing species.
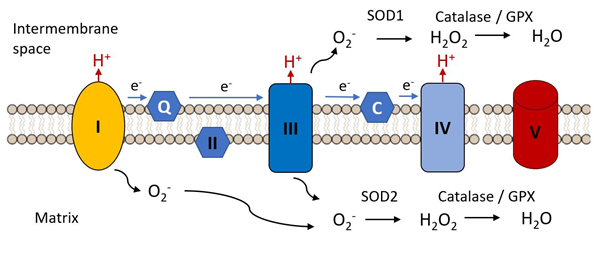
Figure 3. Complex I and III are the major sites of the electron transfer chain at which reactive oxygen species are released. The superoxide radical (.O2−) is the initial species generated by reduction of molecular oxygen. It is reduced to hydrogen peroxide by superoxide dismutases 1 or 2 and is further reduced to water by catalase or glutathione peroxidase, these steps constituting the initial anti-oxidant defense mechanisms.
The Mitochondrial Free Radical Theory of Ageing (MFRTA) was proposed by Harman [51][52] and suggested that increasing accumulation of mutations with age in mtDNA led to defects in mitochondrially encoded components of the electron transport chain, with an associated increase in free radical production. These in turn produced further damage to mtDNA, leading to a self-propelled cycle resulting in eventual catastrophic damage accumulation. If anti-oxidant defenses are impaired with age, this might exacerbate the effect of increased ROS production. In the study of Reutzel et al. [53] reductions in brain catalase and SOD2 were seen at 18 months in mouse, and a similar reduction in anti-oxidant defenses with age was also seen in the rat as suggested by lowered expression of the antioxidant enzymes peroxiredoxin III (Prx III) and superoxide dismutase 2 (SOD2) [54].
Harman’s hypothesis elegantly linked two concepts (1) the known damaging effect of high levels of free radicals on organismal function and (2) the association of increasing accumulation of ROS-associated mtDNA mutations with age. However, many more recent observations have suggested difficulties in the original formulation of this theory. First, several observations suggested that long-lived species do not always demonstrate lower levels of ROS and the accompanying oxidative damage [55]. Second, loss of antioxidant defenses would be predicted to shorten lifespan but this was not found invariably to be the case. Loss of catalase, thought to be an important component of the endogenous anti-oxidant system, at least in Caenorhabditis, has no effect on lifespan [56][57]. and overexpression of catalase, either alone or in combination with superoxide dismutase did not increase lifespan [58]. Third, an increase in ROS signaling may lead to a paradoxical increase in life expectancy [59]. Yang and Hekimi [60] showed that an increase in measurable oxidative damage was without effect on the lifespan of a long lived strain of worms and Schulz et al. [61] showed that impaired glucose availability forced an increase in respiration and ROS generation in Caenorhabditis, but this was accompanied by an increase, rather than a decrease, in lifespan.
The original form of the hypothesis in which oxidative damage caused by mitochondrial dysfunction initiates a vicious cycle of increased ROS production and increasing macromolecular damage culminating in manifestations of ageing and death has been superseded by a more complex model. In this model, which direct damage to macromolecules may be component mechanism, but it is supplemented by equally important epigenetic mechanisms. Thus, Fouquerel et al. [62] showed that selectively targeting the oxidatively modified base 8-oxoguanine to telomeres resulted in telomere shortening and genetic instability, directly linking a product of ROS generation with what is currently believed to be a key element in the ageing process [63].
An indication of the need for subtlety in thinking about the relation of ROS to ageing and lifespan comes from the finding by Bazopoulou et al. [64] that a transient (10 h) increase in ROS in early development of Caenorhabditis triggers epigenetic mechanisms that result in increased stress resistance and redox homeostasis that in turn result in life span extension, the mediating mechanism being global decreases in histone H3K4me3 levels. A biphasic response such as this, where low levels of a potentially damaging molecule which is toxic at higher doses triggers the upregulation of cellular defense mechanisms that are ultimately beneficial to life expectancy was termed mitohormesis by Tapia [65].
Figure 4 indicates the key elements of a revised MFRTA.
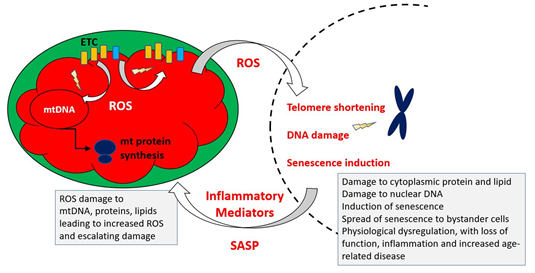
Figure 4. Summary of mechanism proposed by the mitochondrial free radical theory of ageing. Reactive oxygen species produced during operation of the electron transfer chain cause damage to mitochondrial components, including mtDNA, proteins and lipids. This results in accumulation of increasingly damaged components of the electron transport chain (ETC), resulting in exacerbated production of ROS, which are also able to damage extra-mitochondrial cellular components. Additionally, the increased ROS may trigger epigenetic changes, including telomere shortening and upregulation of cellular senescence pathways.
4. Cellular Programs and Aging–Mitochondria in Stem Cells and Senescence
Stem cell function is impaired with age and is responsible for many of the obvious hallmarks of normal ageing such as sarcopenia, decreased immune function and slower wound healing [66]. Mitochondria have been shown to play a role in determining the fate of stem cells by mechanisms including energetics [67], changes in mitochondrial dynamics [68] or the mitochondrial unfolded protein response [69]. The role of Kreb’s cycle intermediates as metabolic regulators has already been discussed and such regulation also impinges on stem cell fate decisions [70]. Stem cells are typically glycolytic with a low abundance of mitochondria [71] and several types of adult stem cell including neural and hematopoietic require a shift to OXPHOS in order to differentiate [72][73]. A declining ability of mitochondria to carry out efficient OXPHOS may therefore have an impact on one key aspect of ageing, the inability effectively to replenish stem cell pools and produce specialized post mitotic cell types in various organ systems [74]. Katajisto et al. [74] followed the fates of old and young mitochondria during the division of human mammary stem like cells and found that such cells apportion aged mitochondria asymmetrically between daughter cells. Daughter cells that received fewer old mitochondria maintained stem cell traits and inhibition of mitochondrial fission disrupted the age-dependent subcellular segregation of mitochondria and caused loss of stem cell properties in the progeny cells.
In addition to loss of stem cell self-renewal and potency, another aspect of ageing is the accumulation in many tissues of populations of post-mitotic and non-functional senescent cells. In spite of their possibly beneficial roles in some specific circumstances (development, cancer prevention and wound healing), the accumulation of senescent cells with age has a deleterious effect on organ function in a range of tissues. This is partly due to their loss of differentiated function, as they contribute to tissue mass without performing tissue specific roles. In addition, they release a complex pro-inflammatory mixture of cytokines, growth factors and enzymes (the senescence associated secretory phenotype, SASP) that can drive bystander cells into senescence and contribute to the damaging inflammation that accompanies ageing [75][76][77][78][79][80]—see Figure 9. Senescent cells are themselves a source of ROS as a result of their dysfunctional mitochondria [81] and ROS-dependent feedback loops can positively reinforce the senescence pathway [81][82].
Defective mitochondria are also linked to the induction of senescence via their role in triggering the SASP. Mitochondrial DNA is normally absent from the cytoplasm of healthy cells, as defective mitochondria are dealt with by mitophagy, in which the entire organelle is enclosed by a phagocytic membrane and the contents are degraded. This prevents exposure of the cytoplasm to mtDNA. However, under conditions of cellular stress or when mitophagic mechanisms are insufficient, damaged mitochondria can release DNA, which is sensed as a damage associated molecular signal by the intracellular enzyme cGAS [83]. This in turn signals via the gene regulator STING to activate IRF3 and NFκB regulated proinflammatory genes. This mix is able to induce senescence in bystander cells. These processes are illustrated in Fig 5. A general increase in circulating mtDNA after the fifth decade was reported by Pinti et al. [84]. Although Jylhävä et al. [85] did not find an association between circulating mtDNA and age, they did find that levels of mitochondrial DNA were correlated with increasing frailty.
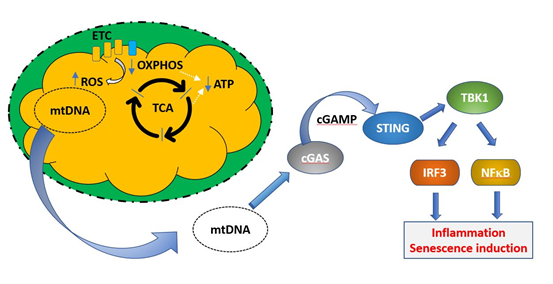
Figure 5. Induction of cellular senescence programs by mitochondrial DNA. Under normal healthy conditions, defective mitochondria (exhibiting decreased OXPHOS, blue arrow, decreased ATP production, green arrow, and increased ROS production, red arrow) are removed by mitophagy and mitochondrial DNA is not released into the cytoplasm. If this process is unable to deal with the increasing load of mitochondrial damage during ageing, mtDNA may be released to the cytoplasm, where it constitutes a damage associated molecular pattern (DAMP). mtDNA is recognized by the enzyme cGAMP, which synthesizes the intermediate cyclic guanosine monophosphate–adenosine monophosphate (cGAMP). This activates the transcription factor stimulator of interferon genes (STING), which upregulates transcriptional programs controlled by IRF3 and NFκB, resulting in the induction of cellular senescence.
5. Mitochondrial Quality Control
Balanced control of mitochondrial dynamics through the processes of fission and fusion, in addition to regulation of the selective removal of damaged mitochondria by mitophagy are essential to mitochondrial quality control. Their importance is indicated by the fact that impairment of any of them is associated with poor cell function and viability, and altered morphology of the network towards either a highly fragmented or a hyperfused morphology is associated with disease and ageing. Key elements of this system are the proteins mediating mitochondrial fission, Drp1 and its mitochondrial outer membrane receptors, mitochondrial fission factor (MFF), fission protein-1 (FIS1), mitochondrial dynamics protein-49 (MiD49) and mitochondrial dynamics protein-51 (MiD51) [86][87], and those mediating fusion the best characterized of which are Optic Atrophy 1 (OPA1) on the inner membrane and Mitofusins 1 and 2 (Mfn1, 2) on the outer membrane [88].
Age related changes in both the components of the dynamics machinery and in the balance between fission and fusion have been described in numerous organisms, the bulk of the evidence in lower organisms tending to suggest that it is the balance between fission and fusion that is most critical to health and longevity. Thus in C. elegans, pathologies associated with inhibition of one process can be reversed by simultaneous inhibition of the other [89]. However, in Saccharomyces cerevisiae, although double mutants lacking the genes dnm1 and deficient for both fission and fusion appear to have normal wild type mitochondrial morphology, they have a shorter lifespan, suggesting that the adynamic condition itself is maladaptive. This phenotype was particularly marked under conditions of nutrient stress, suggesting impaired ability to adjust mitochondrial metabolism to prevailing conditions [90].
Mitophagy is an evolutionary conserved pathway and primarily serves a housekeeping role by recycling mitochondria and adjusting the pool to respond to cellular needs. It can also act as stress response pathway that selectively marks and eliminates damaged mitochondria in order to maintain a healthy mitochondrial population [91]. Several publications provide evidence indicating that aging tissues display an accelerated decline in mitophagy efficiency and an aberrant accumulation of damaged mitochondria that affect health and lifespan in different organisms [92][93]. Building on earlier work in which cardiac ablation of either Drp1 or Mfn1/Mfn2 produced extensive mortality by six weeks of age, Song et al. [94] showed that simultaneous knockout of all three proteins mitigated this pathology to a less aggressive cardiac hypertrophy. These observations support the conclusion that an inability to perform dynamic fission/fusion is itself maladaptive and also provide an important caveat to assumptions that fragmented mitochondrial networks are necessarily dysfunctional.
6. Conclusions
The free radical theory of mitochondrial ageing was the most comprehensive attempt to attribute such a causal role to the mitochondrion but its original formulation has lost support as the complex effects of ROS on ageing have become better appreciated. Similarly, the finding that a deleterious mitochondrial mutation must achieve approximately 60% heteroplasmy has made simple genetic models of mutation untenable. Several additional processes have been identified which may link mitochondrial biology with ageing. These include epigenetic rather than directly damaging effects of ROS, possibly linking ROS to telomere shortening, which is one of the best substantiated biochemical processes controlling cellular lifespan. Effects of mitochondrial function on cellular properties such as senescence and stemness are also likely to provide meaningful links, while a decline in the efficacy of the dynamic mechanisms regulating mitochondrial quality have also been implicated. The complexity of these processes and their inter relationships are still not fully understood and at this point it seems unlikely that a single linear cause and effect relationship between any specific aspect of mitochondrial biology and ageing can be established in either direction. It is probably more useful to think in terms of an interlocking web of mutually reinforcing relationships and in this sense indeed, mitochondrial function and ageing are intimate relations.
References
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta 1999, 1410, 103–123.
- Chinnery, P.F.; Hudson, G. Mitochondrial genetics. Br. Med. Bull. 2013, 106, 135–159.
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–15623.
- Stauch, K.L.; Purnell, P.R.; Fox, H.S. Aging synaptic mitochondria exhibit dynamic proteomic changes while maintaining bioenergetic function. Aging (Albany NY) 2014, 6, 320–334.
- Zahn, J.M.; Sonu, R.; Vogel, H.; Crane, E.; Mazan-Mamczarz, K.; Rabkin, R.; Davis, R.W.; Becker, K.G.; Owen, A.B.; Kim, S.K. Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genet. 2006, 2, e115.
- Terman, A. Catabolic insufficiency and aging. Ann. NY Acad. Sci. 2006, 1067, 27–36.
- Beckman, K.B.; Ames, B.N. Endogenous oxidative damage of mtDNA. Mutat. Res. 1999, 424, 51–58.
- Radzvilavicius, A.L.; Hadjivasiliou, Z.; Pomiankowski, A.; Lane, N. Selection for Mitochondrial Quality Drives Evolution of the Germline. PLoS Biol. 2016, 14, e2000410.
- Ziada, A.S.; Lu, M.Y.; Ignas-Menzies, J.; Paintsil, E.; Li, M.; Ogbuagu, O.; Saberi, S.; Hsieh, A.Y.Y.; Sattha, B.; Harrigan, P.R.; et al. Mitochondrial DNA somatic mutation burden and heteroplasmy are associated with chronological age, smoking, and HIV infection. Aging Cell 2019, 18, e13018.
- Reichart, G.; Mayer, J.; Tokay, T.; Lange, F.; Johne, C.; Baltrusch, S.; Tiedge, M.; Fuellen, G.; Ibrahim, S.; Köhling, R. Combination of mitochondrial tRNA and OXPHOS mutation reduces lifespan and physical condition in aged mice. bioRxiv 2017, 233593, doi:10.1101/233593.
- Fan, W.; Evans, R. PPARs and ERRs: Molecular mediators of mitochondrial metabolism. Curr. Opin. Cell Biol. 2015, 33, 49–54.
- Li, F.; Wang, Y.; Zeller, K.I.; Potter, J.J.; Wonsey, D.R.; OʼDonnell, K.A.; Kim, J.W.; Yustein, J.T.; Lee, L.A.; Dang, C.V. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell Biol. 2005, 25, 6225–6234.
- Dhar, S.S.; Ongwijitwat, S.; Wong-Riley, M.T. Nuclear respiratory factor 1 regulates all ten nuclear-encoded subunits of cytochrome c oxidase in neurons. J. Biol. Chem. 2008, 283, 3120–3129.
- Salminen, A.; Kaarniranta, K. AMP- activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012, 11, 230–241.
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Age-related changes in AMPK activation: Role for AMPK phosphatases and inhibitory phosphorylation by upstream signaling pathways. Ageing Res. Rev. 2016, 28, 15–26.
- Qiang, W.; Weiqiang, K.; Qing, Z.; Pengju, Z.; Yi, L. Aging impairs insulin-stimulated glucose uptake in rat skeletal muscle via suppressing AMPKalpha. Exp. Mol. Med. 2007, 39, 53543.
- Reznick, R.M.; Zong, H.; Li, J.; Morino, K.; Moore, I.K.; Yu, H.J.; Liu, Z.X.; Dong, J.; Mustard, K.J.; Hawley, S.A.; et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007, 5, 151–156.
- Beyer, R.E.; Burnett, B.A.; Cartwright, K.J.; Edington, D.W.; Falzon, M.J.; Kreitman, K.R.; Kuhn, T.W.; Ramp, B.J.; Rhee, S.Y.; Rosenwasser, M.J.; et al. Tissue coenzyme Q (ubiquinone) and protein concentrations over the life span of the laboratory rat. Mech. Ageing Dev. 1985, 32, 267–281.
- Kalen, A.; Appelkvist, E.L.; Dallner, G. Age-related changes in the lipid compositions of rat and human tissues. Lipids 1989, 24, 579–584.
- Mailloux, R.J.; Beriault, R.; Lemire, J.; Singh, R.; Chenier, D.R.; Hamel, R.D.; Appanna, V.D. The tricarboxylic acid cycle, an ancient metabolic network with a novel twist. PLoS ONE 2007, 2, e690.
- Zdzisinska, B.; Zurek, A.; Kandefer-Szerszen, M. Alpha-Ketoglutarate as a Molecule with Pleiotropic Activity: Well-Known and Novel Possibilities of Therapeutic Use. Arch. Immunol. Ther. Exp. (Warsz.) 2017, 65, 21–36.
- Salminen, A.; Kauppinen, A.; Hiltunen, M.; Kaarniranta, K. Krebs cycle intermediates regulate DNA and histone methylation: Epigenetic impact on the aging process. Ageing Res. Rev. 2014, 16, 45–65.
- He, W.; Miao, F.J.; Lin, D.C.; Schwandner, R.T.; Wang, Z.; Gao, J.; Chen, J.L.; Tian, H.; Ling, L. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 2004, 429, 188–193.
- Gilissen, J.; Jouret, F.; Pirotte, B.; Hanson, J. Insight into SUCNR1 (GPR91) structure and function. Pharmacol. Ther. 2016, 159, 56–65.
- Hu, J.; Li, T.; Du, X.; Wu, Q.; Le, Y.Z. G protein-coupled receptor 91 signaling in diabetic retinopathy and hypoxic retinal diseases. Vis. Res. 2017, 139, 59–64.
- Zhu, X.H.; Lu, M.; Lee, B.Y.; Ugurbil, K.; Chen, W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc. Natl. Acad. Sci. USA 2015, 112, 2876–2881.
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; DʼAmico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443.
- Mota, R.A.; Sanchez-Bueno, F.; Saenz, L.; Hernandez-Espinosa, D.; Jimeno, J.; Tornel, P.L.; Martinez-Torrano, A.; Ramirez, P.; Parrilla, P.; Yelamos, J. Inhibition of poly(ADP-ribose) polymerase attenuates the severity of acute pancreatitis and associated lung injury. Lab. Investig. 2005, 85, 1250–1262.
- Grube, K.; Burkle, A. Poly(ADP-ribose) polymerase activity in mononuclear leukocytes of 13 mammalian species correlates with species-specific life span. Proc. Natl. Acad. Sci. USA 1992, 89, 11759–11763.
- Muiras, M.L.; Muller, M.; Schachter, F.; Burkle, A. Increased poly(ADP-ribose) polymerase activity in lymphoblastoid cell lines from centenarians. J. Mol. Med. (Berl.) 1998, 76, 346–354.
- Chini, E.N. CD38 as a regulator of cellular NAD: a novel potential pharmacological target for metabolic conditions. Curr. Pharm. Des. 2009, 15, 57–63.
- Amici, S.A.; Young, N.A.; Narvaez-Miranda, J.; Jablonski, K.A.; Arcos, J.; Rosas, L.; Papenfuss, T.L.; Torrelles, J.B.; Jarjour, W.N.; Guerau-De-Arellano, M. CD38 Is Robustly Induced in Human Macrophages and Monocytes in Inflammatory Conditions. Front. Immunol. 2018, 9, 1593.
- Braidy, N.; Poljak, A.; Grant, R.; Jayasena, T.; Mansour, H.; Chan-Ling, T.; Guillemin, G.J.; Smythe, G.; Sachdev, P.S. Mapping NAD(+) metabolism in the brain of ageing Wistar rats: Potential targets for influencing brain senescence. Biogerontology 2014, 15, 177–198.
- Camacho-Pereira, J.; Tarrago, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139.
- Kim, H.J.; Kim, K.W.; Yu, B.P.; Chung, H.Y. The effect of age on cyclooxygenase-2 gene expression: NF-kappaB activation and IkappaBalpha degradation. Free. Radic. Biol. Med. 2000, 28, 683–692.
- Lumeng, C.N.; Liu, J.; Geletka, L.; Delaney, C.; DelProposto, J.; Desai, A.; Oatmen, K.; Martinez-Santibanez, G.; Julius, A.; Garg, S.; et al. Aging Is Associated with an Increase in T Cells and Inflammatory Macrophages in Visceral Adipose Tissue. J. Immunol. 2011, 187, 6208–6216.
- Hearps, A.C.; Martin, G.E.; Angelovich, T.A.; Cheng, W.J.; Maisa, A.; Landay, A.L.; Jaworowski, A.; Crowe, S.M. Aging is associated with chronic innate immune activation and dysregulation of monocyte phenotype and function. Aging Cell 2012, 11, 867–875.
- Jurk, D.; Wilson, C.; Passos, J.F.; Oakley, F.; Correia-Melo, C.; Greaves, L.; Saretzki, G.; Fox, C.; Lawless, C.; Anderson, R.; et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2014, 2, 4172.
- Rodier, F.; Coppé, J.P.; Patil, C.K.; Hoeijmakers, W.A.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979.
- Tarragó, M.G.; Chini, C.C.S.; Kanamori, K.S.; Warner, G.M.; Caride, A.; De Oliveira, G.C.; Rud, M.; Samani, A.; Hein, K.Z.; Huang, R.; et al. A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD+ Decline. Cell Metab. 2018, 27, 1081–1095.e10.
- Schlame, M.; Greenberg, M.L. Biosynthesis, remodeling and turnover of mitochondrial cardiolipin. Biochim. Biophys. Acta 2016, 1, 3–7, doi:10.1016/j.bbalip.2016.08.010.
- Kagan, V.E.; Chu, C.T.; Tyurina, Y.Y.; Cheikhi, A.; Bayir, H. Cardiolipin asymmetry, oxidation and signaling. Chem. Phys. Lipids 2014, 179, 64–69, doi:10.1016/j.chemphyslip.2013.11.010.
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Ruggiero, F.M. The effect of reactive oxygen species generated from the mitochondrial electron transport chain on the cytochrome c oxidase activity and on the cardiolipin content in bovine heart submitochondrial particles. FEBS Lett. 2000, 466, 323–326.
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Ruggiero, F.M. Reactive oxygen species generated by the mitochondrial respiratory chain affect the complex III activity via cardiolipin peroxidation in beef-heart submitochondrial particles. Mitochondrion 2001, 1, 151–159.
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nature 2013, 15, 1197–1205.
- Petrosillo, G.; Casanova, G.; Matera, M.; Ruggiero, F.M.; Paradies, G. Interaction of peroxidized cardiolipin with rat-heart mitochondrial membranes: Induction of permeability transition and cytochrome c release. FEBS Lett. 2006, 580, 6311–6316.
- Schug, Z.T.; Gottlieb, E. Cardiolipin acts as a mitochondrial signaling platform to launch apoptosis. Biochim. Biophys. Acta (BBA) Biomembr. 2009, 1788, 2022–2031.
- Sen, T.; Sen, N.; Jana, S.; Khan, F.H.; Chatterjee, U.; Chakrabarti, S. Depolarization and cardiolipin depletion in aged rat brain mitochondria: Relationship with oxidative stress and electron transport chain activity. Neurochem. Int. 2007, 50, 719–725.
- Vorbeck, M.L.; Martin, A.P.; Long, J.W., Jr.; Smith, J.M.; Orr, R.R., Jr. Aging-dependent modification of lipid composition and lipid structural order parameter of hepatic mitochondria. Arch. Biochem. Biophys. 1982, 217, 351–361.
- Paradies, G.; Ruggiero. F.M.; Petrosillo, G.; Quagliariello, E. Age-dependent decline in the cytochrome c oxidase activity in rat heart mitochondria: Role of cardiolipin. FEBS Lett. 1997, 406, 136–138.
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300.
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147.
- Reutzel, M.; Grewal, R.; Dilberger, B.; Silaidos, C.; Joppe, A.; Eckert, G. P. Cerebral Mitochondrial Function and Cognitive Performance during Aging: A Longitudinal Study in NMRI Mice. Oxid. Med. Cell Longev. 2020, 4060769.
- Cand, F.; Verdetti, J. Superoxide dismutase, glutathione peroxidase, catalase, and lipid peroxidation in the major organs of the aging rats. Free Radic. Biol. Med. 1989, 7, 59–63.
- Lewis, K.N.; Andziak, B.; Yang, T.; Buffenstein, R. The naked mole-rat response to oxidative stress: Just deal with it. Antioxid. Redox Signal. 2013, 19, 1388–1399.
- Yang, W.; Li, J.; Hekimi, S. A Measurable increase in oxidative damage due to reduction in superoxide detoxification fails to shorten the life span of long-lived mitochondrial mutants of Caenorhabditis elegans. Genetics 2007, 177, 2063–2074.
- Van Raamsdonk, J.M.; Hekimi, S. Superoxide dismutase is dispensable for normal animal lifespan. Proc. Natl. Acad. Sci. USA 2012, 109, 5785–5790.
- Doonan, R.; McElwee, J.J.; Matthijssens, F.; Walker, G.A.; Houthoofd, K.; Back, P.; Matscheski, A.; Vanfleteren, J.R.; Gems, D. Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev. 2008, 22, 3236–3241.
- Ristow, M.; Schmeisser, S. Extending life span by increasing oxidative stress. Free Radic. Biol. Med. 2011, 51, 327–336.
- Yang, W.; Hekimi, S. Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell 2010, 9, 433–447.
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007, 6, 280–293.
- Fouquerel, E.; Barnes, R.P.; Uttam, S.; Watkins, S.C.; Bruchez, M.P.; Opresko, P.L. Targeted and Persistent 8-Oxoguanine Base Damage at Telomeres Promotes Telomere Loss and Crisis. Mol. Cell 2019, 75, 117e6–130e6.
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007, 5, e110.
- Bazopoulou, D.; Knoefler, D.; Zheng, Y.; Ulrich, K.; Oleson, B.J.; Xie, L.; Kim, M.; Kaufmann, A.; Lee, Y.T.; Dou, Y.; et al. Developmental ROS individualizes organismal stress resistance and lifespan. Nature 2019, 576, 301–305.
- Tapia, P.C. Sublethal mitochondrial stress with an attendant stoichiometric augmentation of reactive oxygen species may precipitate many of the beneficial alterations in cellular physiology produced by caloric restriction, intermittent fasting, exercise and dietary phytonutrients: “Mitohormesis” for health and vitality. Med. Hypotheses 2006, 66, 832–843.
- Van Zant, G.; Liang, Y. The role of stem cells in aging. Exp. Hematol. 2003, 31, 659–672.
- Anso, E.; Weinberg, S.E.; Diebold, L.P.; Thompson, B.J.; Malinge, S.; Schumacker, P.T.; Liu, X.; Zhang, Y.; Shao, Z.; Steadman, M.; et al. The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat. Cell Biol. 2017, 19, 614–625.
- Khacho, M.; Clark, A.; Svoboda, D.S.; Azzi, J.; MacLaurin, J.G.; Meghaizel, C.; Sesaki, H.; Lagace, D.C.; Germain, M.; Harper, M.E.; et al. Mitochondrial Dynamics Impacts Stem Cell Identity and Fate Decisions by Regulating a Nuclear Transcriptional Program. Cell Stem Cell 2016, 19, 232–247.
- Mohrin, M.; Shin, J.; Liu, Y.; Brown, K.; Luo, H.; Xi, Y.; Haynes, C.M.; Chen, D. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 2015, 347, 1374–1377.
- Williams, N.C.; O'Neill, L.A.J. A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front. Immunol. 2018, 9, 141.
- Rafalski, V.A.; Mancini, E.; Brunet, A. Energy metabolism and energy-sensing pathways in mammalian embryonic and adult stem cell fate. J. Cell Sci. 2012, 125, 5597–608.
- Zheng, X.; Boyer, L.; Jin, M.; Mertens, J.; Kim, Y.; Ma, L.; Ma, L.; Hamm, M.; Gage, F.H.; Hunter, T. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. Elife 2016, 5, e13374.
- Inoue, S.; Noda, S.; Kashima, K.; Nakada, K.; Hayashi, J.; Miyoshi, H. Mitochondrial respiration defects modulate differentiation but not proliferation ofhematopoietic stem and progenitor cells. FEBS Lett. 2010, 584, 3402–3409.
- Katajisto, P.; Döhla, J.; Chaffer, C.L.; Pentinmikko, N.; Marjanovic, N.; Iqbal, S.; Zoncu, R.; Chen, W.W.; Weinberg, R.A.; Sabatini, D.M. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 2015, 348, 340–343.
- Prattichizzo, F.; Giuliani, A.; Recchioni, R.; Bonafè, M.; Marcheselli, F.; De Carolis, S.; Campanati, A.; Giuliodori, K.; Rippo, M.R.; Brugè, F.; et al. Anti-TNF-α treatment modulates SASP and SASP-related microRNAs in endothelial cells and in circulating angiogenic cells. Oncotarget 2016, 7, 11945–11958.
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 2018, 9, 586.
- Hernandez-Segura, A.; Brandenburg, S.; Demaria, M. Induction and Validation of Cellular Senescence in Primary Human Cells. J. Vis. Exp. 2018, 136, 55782.
- Millerand, M.; Berenbaum, F.; Jacques, C. Danger signals and inflammaging in osteoarthritis. Clin. Exp. Rheumatol. 2019, 3, 48–56.
- Nie, L.; Zhang, P.; Wang, Q.; Zhou, X.; Wang, Q. lncRNA—Triggered Macrophage Inflammaging Deteriorates Age-Related Diseases. Mediat. Inflamm. 2019, 2019, 4260309.
- Yoon, Y.S.; Byun, H.O.; Cho, H.; Kim, B.K.; Yoon, G. Complex II defect via down-regulation of iron-sulfur subunit induces mitochondrial dysfunction and cell cycle delay in iron chelation-induce senescence—Associated growth arrest. J. Biol. Chem. 2003, 278, 51577–51586.
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347.
- Zheng, H.; Huang, Q.; Huang, S.; Yang, X.; Zhu, T.; Wang, W.; Wang, H.; He, S.; Ji, L.; Wang, Y.; et al. Senescence Inducer Shikonin ROS-Dependently Suppressed Lung Cancer Progression. Front. Pharmacol. 2018, 9, 519.
- Aarreberg, L.D.; Esser-Nobis, K.; Driscoll, C.; Shuvarikov, A.; Roby, J.A.; Gale, M., Jr. Interleukin-1beta Induces mtDNA Release to Activate Innate Immune Signaling via cGAS-STING. Mol Cell 2019, 74, 801 e6–815 e6.
- Pinti, M.; Cevenini, E.; Nasi, M.; De Biasi, S.; Salvioli, S.; Monti, D.; Benatti, S.; Gibellini, L.; Cotichini, R.; Stazi, M.A.; et al. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur. J. Immunol. 2014, 44, 1552–1162.
- Jylhava, J.; Nevalainen, T.; Marttila, S.; Jylha, M.; Hervonen, A.; Hurme, M. Characterization of the role of distinct plasma cell-free DNA species in age-associated inflammation and frailty. Aging Cell 2013, 12, 388–397.
- Loson, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667.
- Liu, R.; Chan, D.C. The mitochondrial fission receptor Mff selectively recruits oligomerized Drp1. Mol. Biol. Cell 2015, 26, 4466–4477.
- Hall, A.R.; Burke, N.; Dongworth, R.K.; Hausenloy, D.J. Mitochondrial fusion and fission proteins: Novel therapeutic targets for combating cardiovascular disease. Br. J. Pharmacol. 2014, 171, 1890–1906.
- Byrne, J.J.; Soh, M.S.; Chandhok, G.; Vijayaraghavan, T.; Teoh, J.S.; Crawford, S.; Cobham, A.E.; Yapa, N.M.B.; Mirth, C.K.; Neumann, B. Disruption of mitochondrial dynamics affects behaviour and lifespan in Caenorhabditis elegans. Cell Mol. Life Sci. 2019, 76, 1967–1985.
- Bernhardt, D.; Müller, M.; Reichert, A.S.; Osiewacz, H.D. Simultaneous impairment of mitochondrial fission and fusion reduces mitophagy and shortens replicative lifespan. Sci. Rep. 2015, 5, 7885.
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14.
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, II; Holmstrom, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696.
- McWilliams, T.G.; Prescott, A.R.; Allen, G.F.; Tamjar, J.; Munson, M.J.; Thomson, C.; Muqit, M.M.; Ganley, I.G. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 2016, 214, 333–345.
- Song, M.; Franco, A.; Fleischer, J.A.; Zhang, L.; Dorn, G.W. 2nd, Abrogating Mitochondrial Dynamics in Mouse Hearts Accelerates Mitochondrial Senescence. Cell Metab. 2017, 26, 872 e5–883 e5.


