 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rajiv Chaudhary | + 2909 word(s) | 2909 | 2020-10-22 11:31:02 | | | |
| 2 | Rita Xu | -1072 word(s) | 1837 | 2020-10-30 04:20:36 | | |
Video Upload Options
Development of markers for traits related to tolerance to ash dieback and to investigate whether genotypes selected for tolerance were genetically different from susceptible populations.
1. Introduction
Common ash (Fraxinus excelsior L.) is a broad-leaved tree species of major ecological and economical importance in European forests [1]. The species is predominantly distributed throughout northern and central Europe. Common ash is currently suffering from the ash dieback disease epidemic caused by the ascomycete fungus Hymenoscyphus fraxineus (T. Kowalski) Baral, Queloz and Hosoya [2][3], which is an alien invasive fungus pathogen in Europe that affects trees of all ages and has resulted in severe mortality throughout the natural distribution range of common ash [4][5][6]. Ash dieback has had a devastating impact on ash population since first noted in Sweden in 2001 [7][8]. This has resulted in the declaration of common ash as a threatened species in Sweden [9]. The loss of a high proportion of the ash trees will reduce the genetic diversity in the species, possibly making it more vulnerable to other disturbances and cause adverse ecological effects including reduced associated biodiversity [3][4][10][11][12].
Susceptibility to ash dieback disease, often scored as a high level of crown damage, has a strong negative effect on the growth and survival of ash trees [13][14]. Previous studies reported significant levels of phenotypic and genetic variability for susceptibility to ash dieback in common ash in natural forests and established field trials [14][15][16][17]. Although the observed differences in crown damage in natural settings could be due to disease escape, inoculation studies have shown that active defense is involved in the susceptibility phenotype of ash trees [18][19][20]. Susceptibility to ash dieback is a quantitative trait [21][22] and heritability for traits related to ash dieback severity (crown symptoms) is high (narrow sense heritability h2: 0.49–0.53) [13][14][23] (broad sense heritability H2: 0.42–0.54) [16][17], suggesting there is good scope for breeding less susceptible trees in future. Substantial genetic variation together with heritability estimates of susceptibility traits indicate a good potential of selection and breeding of ash genotypes tolerant to ash dieback [12].
Since it takes a long time to evaluate resistance properties against pathogens, partly due to the long generation times in forest trees, phenotyping selection isdifficult, costly and time consuming. Molecular markers can help to identify superior genotypes for tree breeding and restoration. Marker-assisted selection may facilitate early selection of trees with favorable traits and could therefore accelerate the selection process [24]. Harper and co-workers [15] developed molecular markers to identify individuals with low susceptibility to ash dieback using an associative transcriptomics method based on gene expression variants. These markers were associated with tolerance to ash dieback and indicate a role of the MADS box transcription factor genes. However, only three markers [15] were able to significantly predict tolerance to the disease. These molecular markers were used to identify trees with a reduced susceptibility to ash dieback in Danish, British and Swedish ash populations [22][25] and had a moderate predictive capacity. These results are highly encouraging, but for efficient marker-assisted selection several more markers are needed in addition to the available markers. Stocks et al. [26] showed that single-nucleotide polymorphisms (SNPs) associated with fewer ash dieback symptoms allowed better predictions of genomic estimated breeding values (GEBVs), than randomly selected SNPs in a transpopulation genomic selection approach for ash dieback-tolerant trees, further underlining the need to characterize the genetics underlying tolerant phenotypes.
Next-generation sequencing (NGS) has revolutionized plant and animal research through the generation of molecular markers such as SNPs for high-throughput genotyping [27][28][29]. New NGS methods have allowed the construction of high-density linkage maps [30][31][32] and association genetics studies [33][34] in trees often use approaches for reduced representation sequencing. We have used a multiplex PCR approach for amplicon sequencing [29] with the aim to identify genetic markers for trait-related tolerance to ash dieback, which enables the enrichment of large numbers of amplicons in a single reaction. In multiplex sequencing, each sample is represented by a unique barcode sequence or tag added to the DNA products that are to be sequenced. The sample with the tag determines which sample the read originated from, enabling the assaying of multiple samples in a single sequencing run. After sequencing, the reads are sorted by the detection of the appropriate barcode.
2. Generation of SNP Markers
Out of 1000 randomly selected amplicons, 655 amplicons (66%) were amplified and produced reads. Amplicons with more than 100 reads in total were retained, leaving 567 amplicons to be mapped to the ash genome. The Illumina HiSeq2500 (SNP&SEQ Platform ,ScilifeLab, Uppsala, Sweden) sequencing of 326 individuals generated on an average 0.19 M reads per genotype of which on average 73,834 reads (38.8%) were kept after demultiplexing (Supplementary File 4). The overall mapping rate was 99.81%–97.83% and the variant calling analysis identified 156,735 putative SNPs in 655 scaffolds in the ash genome. The average size of the SNP holding scaffold was 145 kb. These SNPs were filtered at a call rate of 70% among the sample trees and with a MAF of 0.05. A total of 63 SNPs were identified from 40 scaffolds with an average size of 197.5 kb. The SNP data were filtered to one SNP per scaffold. Forty SNPs and 249 genotypes were retained for a marker-trait association analysis. The pattern of physical LD was examined over the scaffolds harboring the 40 SNPs, covering a total of 8 Mb and the length of the included scaffolds ranged from 503 to 1.35 kb. As expected, none of the marker pairs showed complete linkage. The average LD for statistically significant marker pair values in different scaffolds was r2= 0.033 (p < 0.05), and LD estimates were statistically significant for 3.85% of the scaffold marker pairs (p < 0.05) (data not shown).
3. Low Levels of Differentiation between Material Selected for Disease Tolerance Phenotype and Susceptible Wild Population
To examine the potential effects derived from population stratification, we analyzed the population structure using the 40 SNPs both in TASSEL and STRUCTURE v2.3.4. No population structure was observed in the PCA analysis generated in TASSEL. The first two principal components explained 5.8 and 5.5% of the genetic variance in the population. STRUCTURE has also provided some evidence for K = 2 (data not shown) according to the maximum ΔK value (35.4), indicating little population structure in the studied ash population. A low genetic differentiation was detected between tolerant and susceptible genotypes, and genotypes selected for tolerance phenotypes and the rest of the materials (Table 1). A low FST was also observed between the Uppland and Öland population.
Table 1. Pairwise FST comparison of ash population.
|
Type |
Pairwise FST |
|
Tolerant vs. Susceptible genotypes |
0.0220 |
|
Selected for tolerance vs. other materials |
0.0217 |
|
Selected for tolerance vs. Uppland |
0.0228 |
|
Uppland vs. Öland |
0.0187 |
Tolerant: total tolerant ash genotypes; susceptible: total susceptible ash genotypes; selected for tolerance: selected for tolerance phenotypes only, e.g., disease severity; other materials: selected for other traits as wood quality traits (Seed orchard) and unrelated genotypes with disease severity data collected from Uppland and Öland in the year 2016; Uppland: ash genotypes from Uppland; Öland: genotypes from Öland.
4. Marker-Traits Association Identifies Two Scaffolds Associated with the Tolerance Phenotype
To investigate if any association could be detected between the SNPs and the tolerance to ash dieback, we performed marker-traits analyses in TASSEL using an MLM+PCA+K model. The model detected a total of two statistically significant associations (p-value < 0.05) one of which remained significant after correction for multiple testing (FDR < 0.05, Table 2, Figure 1). As a comparison, a GLM+PCA model was also run. This model detected four statistically significant associations between four SNP markers, all of which were identical to the associations with the MLM+PCA+K model and the disease severity of ash (p-value < 0.05, Supplementary File 5a). However, the MLM+PCA+K model showed the smallest departure from the expected p-value (−log10) in the QQ plots, and was therefore least prone to producing false positives. Therefore, the MLM+PCA+K model and its results were considered as the most reliable in the association analysis (Supplementary File 5b).
Table 2. Single-nucleotide polymorphism (SNP) locus annotations and significance values for disease severity in ash at p-value < 0.05 and false discovery rate (FDR) < 0.05 using mixed linear model (MLM).
|
Markera |
Scaffold b |
Variant |
Gene modeld |
Gene model CDS length(Bp) |
Positione |
p-value |
FDR adj p-valuef |
PVE%g |
SNP featureh |
Annotation |
|
SCONTIG5992_29927 |
Scaffold 5992 |
A/G |
FRAEX38873_v2_000299890.1 |
2349 |
29,927 |
0.001 |
0.048 |
5.4 |
non-synonymous |
Peptidase S8, subtilisin-related|Peptidase S8/S53 domain |
|
SCONTIG6368_39377 |
Scaffold 6368 |
C/G |
FRAEX38873_v2_000311990.1 |
2601 |
39,377 |
0.028 |
0.568 |
3.0 |
non-synonymous |
Leucine-rich repeat |
|
SCONTIG2549_5550 |
Scaffold 2549 |
T/A |
FRAEX38873_v2_000132480.1 |
2571 |
5550 |
0.075 |
1.000 |
2.2 |
synonymous |
Pentatricopeptide repeat |
|
SCONTIG8553_81512 |
Scaffold 8553 |
A/G |
FRAEX38873_v2_000368890.1 |
3237 |
81,512 |
0.079 |
0.790 |
2.1 |
non-synonymous |
unknown |
|
SCONTIG874_32729 |
Scaffold 874 |
A/C |
FRAEX38873_v2_000373260.1 |
1464 |
32,729 |
0.089 |
0.716 |
2.1 |
synonymous |
cytochrome p450 94a1-like |
a SNP marker, the SNP name was composed of the scaffold number and SNP position on scaffold; b scaffold name in ash genome; c variation in major and minor allele frequency; d unique gene model in ash genome; e position of SNP in the gene model; fadjusted p-value (false discovery rate) by Benjamini–Hochberg method; g percentage of phenotypic variance explained; h SNP variant.
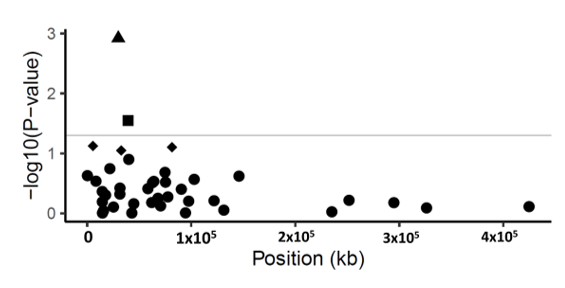
Figure 1. Manhattan plot of p-values (−log10 scale) for SNP associations to disease severity in common ash. The p-values are plotted against position of the SNP in the scaffolds. Grey line indicates the threshold value (−log10 of p-value of 1.3) for declaring a significant association. Triangle symbol indicates the statistically significant SNP (p-value < 0.001, FDR = 0.04) in scaffold 5992; square symbols show marginally significant SNPs (p-value < 0.05). Diamond-shaped symbol indicates non-significant SNPs (p-value > 0.05) and circles are the non-associated SNPs with disease severity.
The marker-trait association generated a statistically significant association with one scaffold. This scaffold (scaffold 5992) comprised five gene models (Supplementary File 6). The SNP SCONTIG5992_29927 on scaffold 5992 was significantly associated to the disease severity of ash (p-value < 0.001, FDR = 0.04), explaining 5.4% of the phenotypic variance (Table 2). This SNP, SCONTIG5992_29927, is located at 29,927 bp in a gene model (FRAEX38873_v2_000299890.1), predicted to encode a Peptidase S8 subtilisin-related Peptidase S8/S53 domain. The substitution encoded by the SNP SCONTIG5992_29927 is located in the coding region changing the amino acid at position 658 in the predicted protein from a tyrosine to an aspartic acid. Another marginally significant SNP (p < 0.05 but FDR > 0.05) SCONTIG5992_29954 was detected on scaffold 5992. This SNP was located 27 bp downstream of SCONTIG5992_29927 in the same gene model (Supplementary File 6). This SNP substitutes the amino acid at position 667 in the predicted protein from an alanine to an isoleucine. Both substitutions are predicted to be buried in the mature protein and located outside the active and catalytic site according to PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/, accessed on 19/10/2019) and I-TASSER (https://zhanglab.ccmb.med.umich.edu/I-TASSER/, accessed on 30/10/2019 ) analyses. The 5.87 kb scaffold 5992 contains four other gene models, of which three are annotated as a pectin methylesterase (FRAEX38873_v2_000299870.1), proteasome subunit alpha type 5 (FRAEX38873_v2_000299880.1), and fyve finger-containing phosphoinositide (FRAEX38873_v2_000299900.1) (Supplementary File 6).
One marginally significant association, i.e., p < 0.05 and FDR > 0.05, was also detected in the marker-trait association, namely SCONTIG6368_39377, in scaffold 6368 contributing to 3.0% of PVE (phenotypic variance explained). This harbors four gene models (Supplementary File 6) and the SNP SCONTIG6368_39377 is located within the gene model FRAEX38873_v2_000311990.1 at position 39,377 bp. This gene model encodes a Leucine-rich repeat protein (Table 2, Figure 1); based on the available predicted protein sequence, this SNP is also non-synonymous and would lead to the substitution of an arginine to a glycine (Table 2, Figure 1).
References
- FRAXIGEN. Ash species in Europe: biological characteristics and practical guidelines for sustainable use. Folia Oecologica 2005, 33 (2), 137.
- Baral, H.O.; Queloz, V.; Hosoya, T. Hymenoscyphus fraxineus, the correct scientific name for the fungus causing ash dieback in Europe. IMA fungus 2014, 5 (1), 79-80.
- Enderle, R.; Stenlid, J.; Vasaitis, R. An overview of ash (Fraxinus spp.) and the ash dieback disease in Europe. CAB Reviews 2019, 14 (025), 1-12.
- Pautasso, M.; Aas, G.; Queloz, V.; Holdenrieder, O. European ash (Fraxinus excelsior) dieback–A conservation biology challenge. Biological conservation 2013, 158, 37-49.
- Coker, T. L.; Rozsypálek, J.; Edwards, A.; Harwood, T. P.; Butfoy, L.; Buggs, R. J. Estimating mortality rates of European ash (Fraxinus excelsior) under the ash dieback (Hymenoscyphus fraxineus) epidemic. Plants, People, Planet 2019, 1 (1), 48-58.
- Pautasso, M.; Schlegel, M.; Holdenrieder, O. Forest Health in a Changing World. Microbial Ecology 2015, 69 (4), 826-842.
- Barklund, P. Askdöd grasserar över Syd-och Mellansverige. SkogsEko 2005, 3, 11-13.
- Cleary, M.; Nguyen, D.; Stener, L.; Stenlid, J.; Skovsgaard, J. Ash and ash dieback in Sweden: A review of disease history, current status, pathogen and host dynamics, host tolerance and management options in forests and landscapes. Dieback of European Ash (Fraxinus spp.): Consequences and Guidelines for Sustainable Management 2017, 195-208.
- Stenlid, J.; Oliva, J.; Boberg, J. B.; Hopkins, A. J. Emerging diseases in European forest ecosystems and responses in society. Forests 2011, 2 (2), 486-504.
- Hultberg, T.; Sandström, J.; Felton, A.; Öhman, K.; Rönnberg, J.; Witzell, J.; Cleary, M. Ash dieback risks an extinction cascade. Biological Conservation 2020, 244, 108516.
- Jönsson, M. T.; Thor, G. Estimating coextinction risks from epidemic tree death: affiliate lichen communities among diseased host tree populations of Fraxinus excelsior. PLoS One 2012, 7 (9), e45701.
- Pliura, A.; Bakys, R.; Suchockas, V.; Marčiulyniene, D.; Gustiene, V.; Verbyla, V.; Lygis, V. Ash dieback in Lithuania: disease history, research on impact and genetic variation in disease resistance, tree breeding and options for forest management. Dieback of European Ash (Fraxinus spp.): Consequences and Guidelines for Sustainable Management 2017, 150-165.
- Kjær, E. D.; McKinney, L. V.; Nielsen, L. R.; Hansen, L. N.; Hansen, J. K. Adaptive potential of ash (Fraxinus excelsior) populations against the novel emerging pathogen Hymenoscyphus pseudoalbidus. Evolutionary applications 2012, 5 (3), 219-228.
- Lobo, A.; Hansen, J. K.; McKinney, L. V.; Nielsen, L. R.; Kjær, E. D. Genetic variation in dieback resistance: growth and survival of Fraxinus excelsior under the influence of Hymenoscyphus pseudoalbidus. Scandinavian Journal of Forest Research 2014, 29 (6), 519-526.
- Harper, A. L.; McKinney, L. V.; Nielsen, L. R.; Havlickova, L.; Li, Y.; Trick, M.; Fraser, F.; Wang, L.; Fellgett, A.; Sollars, E. S.; et al. Molecular markers for tolerance of European ash (Fraxinus excelsior) to dieback disease identified using Associative Transcriptomics. Scientific reports 2016, 6, 19335.
- McKinney, L. V.; Nielsen, L. R.; Hansen, J. K.; Kjær, E. D. Presence of natural genetic resistance in Fraxinus excelsior (Oleraceae) to Chalara fraxinea (Ascomycota): an emerging infectious disease. Heredity 2011, 106 (5), 788.
- Stener, L.G. Clonal differences in susceptibility to the dieback of Fraxinus excelsior in southern Sweden. Scandinavian Journal of Forest Research 2013, 28 (3), 205-216.
- Kjær, E. D.; McKinney, L. V.; Hansen, L. N.; Olrik, D. C.; Lobo, A.; Thomsen, I. M.; Hansen, J. K.; Nielsen, L. R. Genetics of ash dieback resistance in a restoration context–experiences from Denmark. Dieback of European Ash 2017, 106-114.
- Lobo, A.; McKinney, L. V.; Hansen, J. K.; Kjær, E. D.; Nielsen, L. R. Genetic variation in dieback resistance in Fraxinus excelsior confirmed by progeny inoculation assay. Forest pathology 2015, 45 (5), 379-387.
- McKinney, L. V.; Thomsen, I. M.; Kjær, E. D.; Nielsen, L. R. Genetic resistance to Hymenoscyphus pseudoalbidus limits fungal growth and symptom occurrence in Fraxinus excelsior. Forest Pathology 2012, 42 (1), 69-74.
- McKinney, L. V.; Nielsen, L. R.; Collinge, D. B.; Thomsen, I. M.; Hansen, J. K.; Kjær, E. D. The ash dieback crisis: genetic variation in resistance can prove a long‐term solution. Plant Pathology 2014, 63 (3), 485-499.
- Sollars, E. S.; Harper, A. L.; Kelly, L. J.; Sambles, C. M.; Ramirez-Gonzalez, R. H.; Swarbreck, D.; Kaithakottil, G.; Cooper, E. D.; Uauy, C.; Havlickova, L., et al. Genome sequence and genetic diversity of European ash trees. Nature 2017, 541 (7636), 212.
- Pliura, A.; Lygis, V.; Suchockas, V.; Bartkevicius, E. Performance of twenty four European Fraxinus excelsior populations in three Lithuanian progeny trials with a special emphasis on resistance to Chalara fraxinea. Baltic Forestry 2011, 17 (1), 17-34.
- Neale, D. B.; Savolainen, O. Association genetics of complex traits in conifers. Trends in plant science 2004, 9 (7), 325-330.
- Menkis, A.; Bakys, R.; Stein Åslund, M.; Davydenko, K.; Elfstrand, M.; Stenlid, J.; Vasaitis, R. Identifying Fraxinus excelsior tolerant to ash dieback: Visual field monitoring versus a molecular marker. Forest Pathology 2019, e12572.
- Stocks, J. J.; Metheringham, C. L.; Plumb, W. J.; Lee, S. J.; Kelly, L. J.; Nichols, R. A.; Buggs, R. J. A. Genomic basis of European ash tree resistance to ash dieback fungus. Nat. Ecol. Evol. 2019, 3, 1686–1696.
- Baird, N. A.; Etter, P. D.; Atwood, T. S.; Currey, M. C.; Shiver, A. L.; Lewis, Z. A.; Selker, E. U.; Cresko, W. A.; Johnson, E. A. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PloS one 2008, 3 (10), e3376.
- Elshire, R. J.; Glaubitz, J. C.; Sun, Q.; Poland, J. A.; Kawamoto, K.; Buckler, E. S.; Mitchell, S. E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PloS one 2011, 6 (5), e19379.
- Nguyen-Dumont, T.; Pope, B. J.; Hammet, F.; Southey, M. C.; Park, D. J. A high-plex PCR approach for massively parallel sequencing. Biotechniques 2013, 55 (2), 69-74.
- Marchese, A.; Marra, F. P.; Caruso, T.; Mhelembe, K.; Costa, F.; Fretto, S.; Sargent, D. J. The first high-density sequence characterized SNP-based linkage map of olive ('Olea europaea'L. subsp.'europaea') developed using genotyping by sequencing. Australian Journal of Crop Science 2016, 10 (6), 857.
- Wu, D.; Koch, J.; Coggeshall, M.; Carlson, J. The first genetic linkage map for Fraxinus pennsylvanica and syntenic relationships with four related species. Plant molecular biology 2019, 99 (3), 251-264.
- Zhigunov, A. V.; Ulianich, P. S.; Lebedeva, M. V.; Chang, P. L.; Nuzhdin, S. V.; Potokina, E. K. Development of F1 hybrid population and the high-density linkage map for European aspen (Populus tremula L.) using RADseq technology. BMC plant biology 2017, 17 (1), 180.
- Grattapaglia, D.; de Alencar, S.; Pappas, G. Genome-wide genotyping and SNP discovery by ultra-deep Restriction-Associated DNA (RAD) tag sequencing of pooled samples of E. grandis and E. globulus, BMC proceedings, BioMed Central: 2011; p P45.
- Konar, A.; Choudhury, O.; Bullis, R.; Fiedler, L.; Kruser, J. M.; Stephens, M. T.; Gailing, O.; Schlarbaum, S.; Coggeshall, M. V.; Staton, M. E., et al. High-quality genetic mapping with ddRADseq in the non-model tree Quercus rubra. BMC genomics 2017, 18 (1), 417.


