 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | ANTOINE ITALIANO | + 2236 word(s) | 2236 | 2020-09-30 08:47:00 | | | |
| 2 | Bruce Ren | Meta information modification | 2236 | 2020-10-10 10:41:59 | | |
Video Upload Options
Genomic instability is a hallmark of cancer related to DNA damage response (DDR) deficiencies, offering vulnerabilities for targeted treatment. Poly (ADP-ribose) polymerase (PARP) inhibitors (PARPi) interfere with the efficient repair of DNA damage, particularly in tumors with existing defects in DNA repair, and induce synthetic lethality. PARPi are active across a range of tumor types harboring BRCA mutations and also BRCA-negative cancers, such as ovarian, breast or prostate cancers with homologous recombination deficiencies (HRD). Depending on immune contexture, immune checkpoint inhibitors (ICIs), such as anti-PD1/PD-L1 and anti-CTLA-4, elicit potent antitumor effects and have been approved in various cancers types. Although major breakthroughs have been performed with either PARPi or ICIs alone in multiple cancers, primary or acquired resistance often leads to tumor escape. PARPi-mediated unrepaired DNA damages modulate the tumor immune microenvironment by a range of molecular and cellular mechanisms, such as increasing genomic instability, immune pathway activation, and PD-L1 expression on cancer cells, which might promote responsiveness to ICIs. In this context, PARPi and ICIs represent a rational combination.
1. Introduction
Over the past decade, poly(ADP-ribose) polymerase (PARP) inhibitors (PARPi) and monoclonal antibodies that block immune checkpoints, such as programmed cell-death 1 (PD-1) and cytotoxic T lymphocyte antigen 4 (CTL-4), have transformed the treatment of multiple types of cancers. Immune checkpoint inhibitors (ICIs) used as stand-alone therapeutic interventions give rise to durable objective responses in patients affected by a variety of cancers and have been approved for an ever-growing list of malignancies, including melanoma, non-small cell lung carcinoma (NSCLC), renal cell carcinoma, head and neck squamous cell carcinoma (HNSCC), and Hodgkin’s lymphoma [1][2][3][4][5][6][7]. More recently, monotherapy with PARPi as a maintenance strategy showed significant clinical activity in several cancer types harboring germline loss-of-function BRCA mutations such as ovarian, breast and pancreatic cancer [8][9][10][11]. However, despite these substantial advancements in clinical care, the majority of patients receiving either PARPi or ICIs alone do not provide benefit and a rationale to combine these treatments has emerged [12][13].
The groundbreaking success of anticancer immunotherapy is primarily based on the features of cancer cells and their ability to potentially initiate an antitumor immune response. These notable features include gene mutations resulting in abnormal protein expression patterns, such as neoantigens or tumor-associated antigens (TAAs) [14]. TAAs represent self-antigens that are aberrantly expressed or overexpressed in tumor cells, whereas neoantigens refer to non-self-antigens arising as a result of somatic mutation [14][15]. The formation of mutation-derived TAAs and neoantigen, reflecting the mutational burden of the tumor, allow the immune system to recognize tumor cell and initiate the cancer-immunity cycle [16][17]. The subsequent antitumor immune process against neoantigens relies on several steps, including the release and presentation of cancer antigens by antigen-presenting cells (APC), priming and activation of T cells, trafficking and infiltration of T cells, and recognizing and killing cancer cells [18]. However, a subset of cancer cells can escape host immune destruction by impairing one or more steps and result in tumor progression [19]. The role of immunotherapy is to reinvigorate antitumor immune response by disrupting co-inhibitory T cell signaling, transferring additional tumor-specific T cells clones and reshaping the immunosuppressive microenvironment [20][21][22][23]. Several strategies, including ICIs, adoptive T cell transfer, and vaccination, have been put to use in multiple cancers [24][25][26][27]. Nevertheless, due to complex and constantly evolving interactions between cancer cells and the immune system, both primary and acquired resistance with ICIs monotherapy are observed [28][29]. Therefore, combination treatment with ICIs is an attractive strategy to potentiate efficacy and lower resistance.
Recent molecular profiling of DNA damage repair genes has allowed the implementation of novel therapeutic strategies. By interfering with efficient DNA damage repair, the inhibition of PARP that target the base excision repair (BER) pathway leads to insufficient DNA repair, with subsequent unsustainable DNA damage, and thus represents a synthetic lethal therapeutic approach for the treatment of cancers with compromised ability to repair double-strand DNA breaks by homologous recombination (HR), including those with defects in BRCA1/2 [30]. The unrepaired-DNA promotes immune priming through a range of molecular mechanisms and also leads to adaptative upregulation of programmed death ligand 1 (PD-L1) expression[31]. Moreover, PARPi modulates the inflammatory immune microenvironment of tumors and reinstates a productive TH1 immune response [31]. This multifaceted immunological effect of PARPi might be favorable to boost an antitumor immune response and enhance the efficacy of ICIs. In this review, we summarize the basic and translational biology supporting the combined strategy and provide a focus on preclinical studies and ongoing clinical trials of ICIs combined with PARPi, as well as perspectives and potential challenges of this combination strategy.
2. DNA Damage and PARP Inhibition
2.1. Role of PARP in DNA Damage Response
Cells are continuously faced with endogenous and exogenous stress that can ultimately lead to DNA damage. To preserve genomic integrity and prevent emergence of cancer, detection and repair of DNA is a critical process, managed by multiple pathways [32]. DNA single-strand break (SSB) damage is fixed by three main pathways: (1) BER, (2) nucleotide excision repair (NER), and (3) mismatch-repair (MMR). Possibly more dangerous DNA double-strand breaks (DSB) are restored by two additional pathways: (1) HR and (2) non-homologous end joining (NHEJ) [33]. Anomalies observed in DNA damage response (DDR) key genes, such as BRCA1/2 or TP53, are associated with cancer-prone phenotypes [34]. As a consequence, failure in DDR in an accurate and well-timed manner can result in the defective elimination of genome mutations and increases the risk of oncogenesis after established DNA damage events [35]. Depending on the context, cancer cells often harbor a lessened repertoire of DDR signaling competences, rendering them more reliant on a subset of DNA repair pathways and therefore more susceptible to DDR inhibition than normal cells [36].
PARP1/2 enzymes are core DNA-damage sensor and signal transducer in DDR, which bind to DNA breaks and catalyze the synthesis of poly(ADP-ribose)(PAR) chains on target proteins (PARylation) in the vicinity of the DNA break and itself (autoPARylation) [37]. These negatively charged PAR chains promote chromatin remodeling, recruit DNA repair-related protein complex and affect the replication fork progression speed [38][39]. The binding of PARP1 via zinc finger domains to sites of DNA-damage carries a conformational change in the PARP1 proteins and relieves the autoinhibitory interaction between the catalytic domain and helical domain. Then, the PARP1 co-factor nicotinamide (β-NAD+) is used as a substrate at the active site of the enzyme to catalyze the transfer of ADP-ribose moieties onto target proteins. The synthesis of ADP-ribose polymeric chains on proteins in the vicinity of DNA breaks, called PARylation, likely mediates DNA repair by modifying chromatin structure and by localizing DNA repair effectors [40]. Thereafter DNA-damage restore, autoPARylation occur that rapidly dissociate PARP from damage site [41]. The role of PARP has been well identified in BER-mediated SSB repair pathways, as well as other DDR pathways [42].
2.2. The Lethal Synthetic Effect of PARP Inhibitors
2.1.1. Mechanism of action of PARPi
Although the precise mechanism by which PARPi kill tumor cells remains to be fully clarified, the anticancer effect is attributed to catalytic inhibition of PARP that block repair of DNA SSB [43] (Figure 1a). While PARPi is well-tolerated by normal cells, this effect of PARPi is more likely observed in tumor cells with a BRCA-deficient background [43]. As a result of defective enzymatic function induced by PARPi, the accumulation of SSB is subsequently encountered by replication forks and generates potentially lethal DSBs that need to be fixed [43][44]. In normal cells, the accumulation of DSBs are repaired preferentially by HR rather than NHEJ [45]. HR is a high-fidelity repair pathway that utilizes the sister copy of the damaged DNA as a template, leading to the reconstitution of the original sequence [46]. In contrast, NHEJ is intrinsically error-prone, modifies the broken DNA ends, and ligates them together with little or no homology, generating deletions or insertions [47]. However, in some cancer cells lacking BRCA1 or BRCA2, two key tumor suppressor proteins involved in DSB repair by HR, loss of PARP function leads to the accumulation of DSBs that are unrepaired or unsustainably repaired by NHEJ which results in cell death [48]. Based on the discovery of this synthetic lethality between BRCA and PARP, numerous PARPi have been developed, including olaparib, rucaparib, niraparib, talazoparib, and veliparib, which are mainly applied in cancer patients with BRCA1/2 mutations.
Although the greatest efficacy of PARPi has been observed in tumors with BRCA1/2 mutations, accumulating data indicate that synthetic lethality is inadequate to explain the whole antitumor activity. First, the ability of PARPi to inhibit PARP catalytic activity is poorly correlated to its cell-killing ability in HR deficiencies (HRD) cells [49]. In addition, PARPi induces cytotoxicity to a greater extent than PARP depletion [50][51][52]. Furthermore, PARP itself is essential to the cytotoxic effects of PARPi [52]. Actually, these facts may be attributed in part to the PARP trapping potency of PARPi (Figure 1b). Although the precise mechanisms of PARP trapping remains unclear, it has been proposed that PARPi could either prevent the release of PARP1 from DNA by inhibiting autoPARylation [53]. Likewise, PARPi binding to the catalytic site could cause allosteric changes in the PARP1 structure enhancing DNA avidity. The trapping DNA-PARP complex stalls the progress of replication fork and elicit cytotoxic effects primarily through the conversion of unrepaired SSBs into lethal DSBs [43,49]. Moreover, PARPi could also act via the upregulation of NHEJ pathway, which presumably leads to genomic instability and eventual lethality [54]. Finally, PARPi could suppress the role of PARP in reactivating DNA replication forks and cause cell death. Additional studies further demonstrated that loss of other tumor suppressor DNA repair proteins, many of which are involved in HR, such as RAD51, ataxia telengiectasia Rad3-related (ATR), ataxia telangiectasia mutated (ATM), checkpoint kinase 1 (CHK1), checkpoint kinase 2 (CHK2), and partner and localizer of BRCA2 (PALB2) also caused sensitization to PARPi [55]. These results suggested that PARPi might be a useful therapeutic strategy not only for the treatment of BRCA-mutated tumors but also for the treatment of a wider range of non-BRCA-mutated tumors that are inherently HR deficient (HRD) or “BRCAness/HRDness” [56].
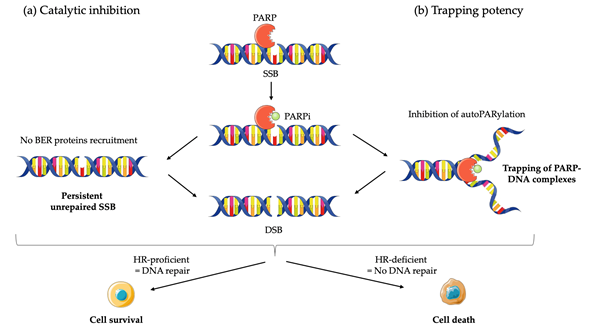
Figure 1. Mechanisms of action of PARP inhibitors (PARPi): (a) PARPi impedes PARP enzyme activity (or catalytic inhibition) and interferes with repair of single strand breaks (SSB) by disrupting the base excision repair (BER) pathway; (b) PARPi also causes trapping of PARP proteins on DNA by inhibiting autoPARylation. In homologous recombination (HR) proficient normal cells, DNA is repair and cell survive. The result in unresolved DNA double strand breaks (DSB) in HR deficient cells leads to cell death.
2.1.2. Clinical Applications of PARPi
The early development of PARPi focused initially on their use in combination with cytotoxic chemotherapy agents and radio-sensitizing drugs, but this was rapidly rejected because of excessive toxicity [57][58][60][61][62]. The potent antitumor effect of PARPi was originally observed in tumors harboring germline BRCA1/2 mutations (gBRCA1/2m), such as familial breast and ovarian cancer [59]. This rapid translation of preclinical studies into promising clinical data triggered the development of several PARPi in different tumors types. Initially, PARPi in the clinic improved clinical benefits for germline or somatic BRCA-deficient ovarian cancer[60][61] . Subsequently, breast, pancreatic and prostate cancers that harbors defects in BRCA also demonstrated to be PARPi responsive[60][61][62]. More recently, it has been suggested that patients without BRCA mutations shared therapeutic vulnerabilities, especially tumors with deficiencies in HR. Indeed, the activity of PARPi is based on the concept of synthetic lethality, where an underlying HRD in tumor cells makes the cells highly susceptible to PARP inhibition. This hypothesis has been further confirmed with multiple clinical studies showing that sensitivity to PARPi occurs in tumors beyond those with BRCA mutations, especially in HRD-positive tumors [63][64][65][66][67]. To date, five PARPi have been approved or orphan drug designed by the FDA (veliparib, rucaparib, talazoparib, niraparib, olaparib) and applied in clinical practice.
Despite the advances of PARPi in a particular population, acquired resistance is a common clinical phenotype. Owing to extensive preclinical studies, several resistance mechanisms have been identified that can be classified into four main categories. Firstly, numerous different mechanisms result in the reactivation of HR function. For example, secondary reversion mutations in several key HR repair (HRR) genes, such as BRCA1/2, RAD51C/D, and PALB2, restore the open reading frame and thus HR competency [68]. Moreover, the loss of p53-binding protein 1 (53BP1), a protein promoting NHEJ, is associated with PARPi resistance by recovery of HRR in BRCA1-deficient tumors [69]. By directly impacting the activity and abundance of PAR chains that decreased PARP trapping, mutations in DNA-binding domains of PARP1 and mechanisms that increase PARylation of PARP1 could also lead to PARPi resistance [70][71]. Furthermore, the cellular availability of the inhibitor is a critical step for successful therapy, as illustrated by the upregulation of ATP-binding cassette (ABC) transporters, such as the P-glycoprotein (PgP) efflux pump that have been described to reduce the efficacy of PARPi [72]. At last, restoration of replication fork protection that induces the stabilization of stalled forks may lead to PARPi resistance [73]. Indeed, fork degradation induced by PARPi is mediated by PTIP and EZH2 proteins, which upon loss lead to protection of the fork from nucleases and thereby resistance [74][75].
Intense preclinical and clinical research are ongoing in order to broadening responding patients, overcoming acquired resistance and enhancing the efficacy of PARPi [73]. The development of combination therapy encompassing PARPi is a potential approach to address these objectives. In addition to the hypothesis that patients with HRD tumors are more prone to produce neoantigen and exhibit higher mutational load, there is a preclinical rational suggesting that PARPi may promote the formation of neoantigen and generate tumor cell recognition by the immune system, making this class of drugs a potential partner for combination with ICIs [76][77].
References
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723.
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. N. Engl. J. Med. 2015, 372, 320–330, doi:10.1056/NEJMoa1412082.
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639, doi:10.1056/NEJMoa1507643.
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833, doi:10.1056/NEJMoa1606774.
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813, doi:10.1056/NEJMoa1510665.
- Ferris, R.L.; Blumenschein, G.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867, doi:10.1056/NEJMoa1602252.
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 Blockade with Nivolumab in Relapsed or Refractory Hodgkin’s Lymphoma. N. Engl. J. Med. 2015, 372, 311–319, doi:10.1056/NEJMoa1411087.
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505, doi:10.1056/NEJMoa1810858.
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA. Mutation. N. Engl. J. Med. 2017, 377, 523–533, doi:10.1056/NEJMoa1706450.
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763, doi:10.1056/NEJMoa1802905.
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA. -Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327, doi:10.1056/NEJMoa1903387.
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355, doi:10.1126/science.aar4060.
- Patel, M.; Nowsheen, S.; Maraboyina, S.; Xia, F. The role of poly(ADP-ribose) polymerase inhibitors in the treatment of cancer and methods to overcome resistance: A review. Cell Biosci. 2020, 10, 35, doi:10.1186/s13578-020-00390-7.
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74, doi:10.1126/science.aaa4971.
- Lee, C.-H.; Yelensky, R.; Jooss, K.; Chan, T.A. Update on Tumor Neoantigens and Their Utility: Why It Is Good to Be Different. Trends Immunol. 2018, 39, 536–548, doi:10.1016/j.it.2018.04.005.
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10, doi:10.1016/j.immuni.2013.07.012.
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608, doi:10.1158/1535-7163.MCT-17-0386.
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer–immune set point. Nature 2017, 541, 321–330, doi:10.1038/nature21349.
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570.
- Granier, C.; De Guillebon, E.; Blanc, C.; Roussel, H.; Badoual, C.; Colin, E.; Saldmann, A.; Gey, A.; Oudard, S.; Tartour, E. Mechanisms of action and rationale for the use of checkpoint inhibitors in cancer. ESMO Open 2017, 2, e000213, doi:10.1136/esmoopen-2017-000213.
- Yu, S.; Li, A.; Liu, Q.; Li, T.; Yuan, X.; Han, X.; Wu, K. Chimeric antigen receptor T cells: A novel therapy for solid tumors. J. Hematol. Oncol. 2017, 10, 78, doi:10.1186/s13045-017-0444-9.
- Marin-Acevedo, J.A.; Dholaria, B.; Soyano, A.E.; Knutson, K.L.; Chumsri, S.; Lou, Y. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J. Hematol. Oncol. 2018, 11, 39, doi:10.1186/s13045-018-0582-8.
- Fucá, G.; Reppel, L.; Landoni, E.; Savoldo, B.; Dotti, G. Enhancing Chimeric Antigen Receptor T cell Efficacy in Solid Tumors. Clin. Cancer Res. 2020, 26, 2444–2451, doi:10.1158/1078-0432.CCR-19-1835.
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422, doi:10.1056/NEJMoa1001294.
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73, doi:10.1056/NEJMra1706169.
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39, doi:10.1016/j.intimp.2018.06.001.
- Lopes, A.; Vandermeulen, G.; Préat, V. Cancer DNA vaccines: Current preclinical and clinical developments and future perspectives. J. Exp. Clin. Cancer Res. 2019, 38, 146, doi:10.1186/s13046-019-1154-7.
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723, doi:10.1016/j.cell.2017.01.017.
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16, doi:10.1038/bjc.2017.434.
- Ashworth, A. A synthetic lethal therapeutic approach: Poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol. 2008, 26, 3785–3790, doi:10.1200/JCO.2008.16.0812.
- Stewart, R.A.; Pilié, P.G.; Yap, T.A. Development of PARP and Immune-Checkpoint Inhibitor Combinations. Cancer Res. 2018, 78, 6717–6725, doi:10.1158/0008-5472.CAN-18-2652.
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 2016, 16, 35–42, doi:10.1038/nrc.2015.4.
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078, doi:10.1038/nature08467.
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204, doi:10.1016/j.molcel.2010.09.019.
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656, doi:10.1016/j.cell.2017.01.002.
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov. 2017, 7, 20–37, doi:10.1158/2159-8290.CD-16-0860.
- De Vos, M.; Schreiber, V.; Dantzer, F. The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochem. Pharmacol. 2012, 84, 137–146, doi:10.1016/j.bcp.2012.03.018.
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158.
- Maya-Mendoza, A.; Moudry, P.; Merchut-Maya, J.M.; Lee, M.; Strauss, R.; Bartek, J. High speed of fork progression induces DNA replication stress and genomic instability. Nature 2018, 559, 279–284, doi:10.1038/s41586-018-0261-5.
- Langelier, M.-F.; Eisemann, T.; Riccio, A.A.; Pascal, J.M. PARP family enzymes: Regulation and catalysis of the poly(ADP-ribose) posttranslational modification. Curr. Opin. Struct. Biol. 2018, 53, 187–198, doi:10.1016/j.sbi.2018.11.002.
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424, doi:10.1038/nrm3376.
- Min, A.; Im, S.-A. PARP Inhibitors as Therapeutics: Beyond Modulation of PARylation. Cancers 2020, 12, 394, doi:10.3390/cancers12020394.
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393, doi:10.1016/j.molonc.2011.07.001.
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921, doi:10.1038/nature03445.
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902–2906, doi:10.4161/cc.7.18.6679.
- Thompson, L.H.; Schild, D. Homologous recombinational repair of DNA ensures mammalian chromosome stability. Mutat. Res. 2001, 477, 131–153, doi:10.1016/s0027-5107(01)00115-4.
- Lieber, M.R. The mechanism of human nonhomologous DNA end joining. J. Biol. Chem. 2008, 283, 1–5, doi:10.1074/jbc.R700039200.
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917, doi:10.1038/nature03443.
- Murai, J.; Huang, S. -y. N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599, doi:10.1158/0008-5472.CAN-12-2753.
- Ström, C.E.; Johansson, F.; Uhlén, M.; Szigyarto, C.A.-K.; Erixon, K.; Helleday, T. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 2011, 39, 3166–3175, doi:10.1093/nar/gkq1241.
- Wang, Y.-Q.; Wang, P.-Y.; Wang, Y.-T.; Yang, G.-F.; Zhang, A.; Miao, Z.-H. An Update on Poly(ADP-ribose)polymerase-1 (PARP-1) Inhibitors: Opportunities and Challenges in Cancer Therapy. J. Med. Chem. 2016, 59, 9575–9598, doi:10.1021/acs.jmedchem.6b00055.
- Shen, Y.; Aoyagi-Scharber, M.; Wang, B. Trapping Poly(ADP-Ribose) Polymerase. J. Pharmacol. Exp. Ther. 2015, 353, 446–457, doi:10.1124/jpet.114.222448.
- Hopkins, T.A.; Shi, Y.; Rodriguez, L.E.; Solomon, L.R.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Wilsbacher, J.L.; Gao, W.; Olson, A.M.; et al. Mechanistic Dissection of PARP1 Trapping and the Impact on In Vivo Tolerability and Efficacy of PARP Inhibitors. Mol. Cancer Res. 2015, 13, 1465–1477, doi:10.1158/1541-7786.MCR-15-0191-T.
- Patel, A.G.; Sarkaria, J.N.; Kaufmann, S.H. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3406–3411, doi:10.1073/pnas.1013715108.
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006, 66, 8109–8115, doi:10.1158/0008-5472.CAN-06-0140.
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120, doi:10.1038/nrc.2015.21.
- Oza, A.M.; Cibula, D.; Benzaquen, A.O.; Poole, C.; Mathijssen, R.H.J.; Sonke, G.S.; Colombo, N.; Špaček, J.; Vuylsteke, P.; Hirte, H.; et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: A randomised phase 2 trial. Lancet Oncol. 2015, 16, 87–97, doi:10.1016/S1470-2045(14)71135-0.
- Dhawan, M.S.; Bartelink, I.H.; Aggarwal, R.R.; Leng, J.; Zhang, J.Z.; Pawlowska, N.; Terranova-Barberio, M.; Grabowsky, J.A.; Gewitz, A.; Chien, A.J.; et al. Differential Toxicity in Patients with and without DNA Repair Mutations: Phase I Study of Carboplatin and Talazoparib in Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 6400–6410, doi:10.1158/1078-0432.CCR-17-0703.
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors from BRCA Mutation Carriers. N. Engl. J. Med. 2009, 361, 123–134, doi:10.1056/NEJMoa0900212.
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164, doi:10.1056/NEJMoa1611310.
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284, doi:10.1016/S1470-2045(17)30469-2.
- Diéras, V.C.; Han, H.S.; Kaufman, B.; Wildiers, H.; Friedlander, M.; Ayoub, J.-P.; Puhalla, S.L.; Bondarenko, I.; Campone, M.; Jakobsen, E.H.; et al. Phase III study of veliparib with carboplatin and paclitaxel in HER2-negative advanced/metastatic gBRCA-associated breast cancer. Ann. Oncol. 2019, 30, v857–v858, doi:10.1093/annonc/mdz394.008.
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708, doi:10.1056/NEJMoa1506859.
- Ledermann, J.A.; Pujade-Lauraine, E. Olaparib as maintenance treatment for patients with platinum-sensitive relapsed ovarian cancer. Ther. Adv. Med. Oncol. 2019, 11, doi:10.1177/1758835919849753.
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428, doi:10.1056/NEJMoa1911361.
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961, doi:10.1016/S0140-6736(17)32440-6.
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415, doi:10.1056/NEJMoa1909707.
- Pettitt, S.J.; Lord, C.J. Dissecting PARP inhibitor resistance with functional genomics. Curr. Opin. Genet. Dev. 2019, 54, 55–63, doi:10.1016/j.gde.2019.03.001.
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.; Ji, J.; Aly, A.; et al. Loss of 53BP1 Causes PARP Inhibitor Resistance in Brca1-Mutated Mouse Mammary Tumors. Cancer Discov. 2013, 3, 68–81, doi:10.1158/2159-8290.CD-12-0049.
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849, doi:10.1038/s41467-018-03917-2.
- Gogola, E.; Duarte, A.A.; de Ruiter, J.R.; Wiegant, W.W.; Schmid, J.A.; de Bruijn, R.; James, D.I.; Guerrero Llobet, S.; Vis, D.J.; Annunziato, S.; et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell. 2018, 33, 1078–1093.e12, doi:10.1016/j.ccell.2018.05.008.
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.H.; Zander, S.A.L.; Derksen, P.W.B.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084, doi:10.1073/pnas.0806092105.
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834, doi:10.1016/j.tcb.2019.07.008.
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.-E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387, doi:10.1038/nature18325.
- Rondinelli, B.; Gogola, E.; Yücel, H.; Duarte, A.A.; van de Ven, M.; van der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378, doi:10.1038/ncb3626.
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.-K.; Hsu, J.-M.; Hsu, J.L.; Yu, W.-H.; Du, Y.; Lee, H.-H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720, doi:10.1158/1078-0432.CCR-16-3215.
- Chabanon, R.M.; Muirhead, G.; Krastev, D.B.; Adam, J.; Morel, D.; Garrido, M.; Lamb, A.; Hénon, C.; Dorvault, N.; Rouanne, M.; et al. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J. Clin. Investig. 2019, 129, 1211–1228, doi:10.1172/JCI123319.


