 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | James Hodgkinson | + 2820 word(s) | 2820 | 2020-09-29 10:27:40 | | | |
| 2 | Catherine Yang | Meta information modification | 2820 | 2020-10-12 10:19:35 | | |
Video Upload Options
Histone deacetylase (HDAC) enzymes play crucial roles in epigenetic gene expression and are an attractive therapeutic target. Five HDAC inhibitors have been approved for cancer treatment to date, however clinical applications have been limited due to poor single agent drug efficacy and side effects associated with a lack of HDAC isoform or complex selectivity. An emerging strategy aiming to address these limitations is the development of bifunctional HDAC therapeutics – single molecules comprising a HDAC inhibitor conjugated to another specificity targeting moiety. There has been many recent advancements in novel types of dual-targeting HDAC modulators, including PROTACs, often achieving high HDAC isoform selectivity, as well as some dual inhibitor examples affording HDAC complex selectivity. Such bifunctional molecules have future potential in achieving enhanced drug efficacy and therapeutic benefits in treating disease.
1. Introduction
The reversible acetylation and deacetylation of protein substrates play critical roles in the regulation of epigenetic gene expression and other cellular processes [1][2][3][4]. These modifications are controlled by two opposing families of enzymes: histone acetyltransferases (HATs) and histone deacetylases (HDACs). In nucleosomal histone tail regions, HDACs catalyse the hydrolysis of N-ε-acetyl-l-lysine side chains to afford acetate and free l-lysine (Figure 1a), resulting in a more compacted chromatin structure which prevents transcription factors and RNA polymerase from accessing gene promoter regions—hence, HDACs are widely associated with gene repression. In addition to histones, HDACs are also responsible for the deacetylation of lysine residues in other proteins, including α-tubulin, heat shock protein 90 (Hsp90), as well as a variety of transcription factors and DNA repair proteins [5]. There are 18 different HDACs present in the human genome. These are divided into two distinct families based on their requirements for activity, as well as four classes based on their sequence homology [6][7]. The more widely explored family of HDACs is the zinc-dependent family, comprising of 11 isoforms divided into 3 classes: class-I (HDACs 1, 2, 3, and 8), class-II (further subdivided into IIa: HDACs 4, 5, 7, and 9, plus IIb: HDACs 6 and 10), and class-IV (HDAC11). The second family requires the cofactor NAD+ for activity as opposed to zinc and encompass the structurally and mechanistically unrelated class-III HDACs, also known as sirtuins (SIRT1-7).
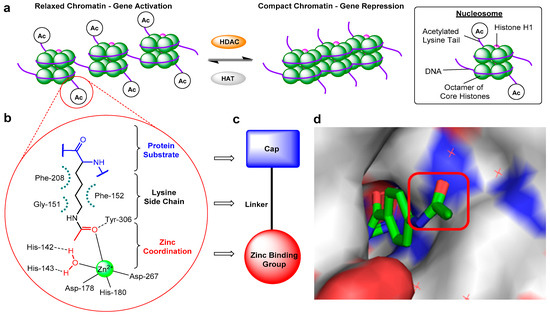
Figure 1. (a) Schematic diagram illustrating changes in chromatin structure due to histone deacetylase (HDAC)-catalysed deacetylation. (b) Representative HDAC–acetyl lysine substrate interactions in an active site. (c) Typical HDAC inhibitor design. (d) Crystal structure of o-aminoanilide HDAC inhibitor bound to HDAC2, highlighting the surface-exposed acetyl group and hence cap modification tolerance for dual inhibitor functionalisation (PDB 4LY1).
Abnormal changes in HDAC expression and therefore the levels of deacetylation have been associated with a range of diseases, including many cancers [8]. For example, HDACs have been shown to influence the expression of numerous genes in both cancer initiation and progression, plus play an essential role in many signalling pathways that promote malignant cell survival [9][10]. Consequently, pharmacological targeting of HDACs has emerged as an important therapeutic area of research, with the discovery of HDAC inhibitors for the treatment of cancers such as leukaemias and myelodysplastic disorders [11], as well as Alzheimer’s disease [12], Huntington’s disease [13], muscular atrophy [14] and Friedrich’s ataxia [15].
Despite mediating the acetylation status of various proteins, the zinc-dependent HDACs possess a mostly conserved catalytic active site (Figure 1b) [16][17], hence the majority of synthesised HDAC inhibitors encompass a generic three-part “cap-linker-zinc binding group” pharmacophore model (Figure 1c) [18]. The crucial aspect of the design is the zinc chelator, which functions by inserting into the HDAC active site and binding zinc at the bottom of the enzyme pocket, usually in a bidentate approach. Next is the linker section, which mimics the substrate lysine side chain by fitting the 11 Å tube-like channel leading to the zinc ion, plus maintaining the multiple hydrophobic interactions within the active site. The final component is the hydrophobic “cap” at the end of the linker, which often contains aromatic moieties that interact with residues near the outer region of the active site or on the protein external surface. The terminal cap points out towards solvent and is routinely optimised to afford inhibitors with HDAC isoform selectivity (Figure 1d).
There are currently four main structural classes of HDAC inhibitors; hydroxamic acids, ortho-aminoanilides, cyclic peptides and aliphatic acids (Figure 2) [19]. The inhibitors are categorised based on their zinc-binding group; however, many structural similarities are seen between the classes. The hydroxamic acids are generally broad-range inhibitors (target HDACs 1–11) and potent at low nM concentrations, causing cell cycle arrest and apoptosis in cultured cells [20]. Conversely, the aliphatic acids are much weaker inhibitors, although, they do exhibit modest class-I HDAC selectivity. The o-aminoanilides, also referred to as benzamides, exhibit high class-I selectivity, as do the cyclic peptides [21].
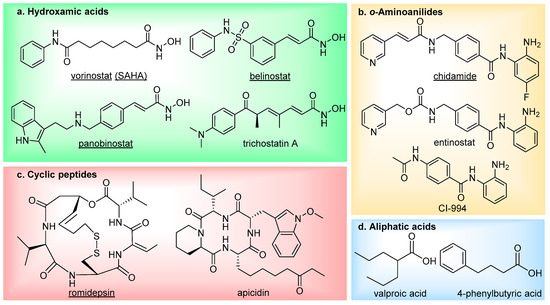
Figure 2. The main four classes of HDAC inhibitors, including FDA-approved drugs (underlined).
To date, five HDAC inhibitors have been approved for cancer treatment-vorinostat and romidepsin to treat refractory cutaneous T-cell lymphoma [22][23], panobinostat for patients with multiple myeloma [24], plus belinostat and chidamide for the treatment of relapsed or refractory peripheral T-cell lymphoma [25][26]. These and additional HDAC inhibitors have also entered clinical trials for the treatment of other types of cancer [27] (NCT00138203, NCT00077194, NCT00451035, NCT00828854, NCT02236195), as well as non-oncology indications such as neurodegenerative diseases (NCT02124083, NCT00212316) and epilepsy (NCT03894826). Although first generation HDAC inhibitors have demonstrated therapeutic significance, especially in treating hematologic malignancies, there has been limitations both in their selectivity and efficacy which has restricted further clinical applications. They frequently suffer from poor HDAC isoform selectivity, resulting in high toxicity and severe side effects [28][29][30][31][32][33]. In addition to this, HDAC inhibitors have also been shown to be less potent against solid tumours, thus hindering employment in cancer treatment [34][35][36][37][38].
2. HDAC Proteolysis-Targeting Chimeras (PROTACs)
A novel approach utilising bifunctional HDAC inhibitors has been in the development of proteolysis-targeting chimeras (PROTACs) to target HDAC enzymes for induced protein degradation. These compounds comprise of three components: a HDAC inhibitor, an E3 ligase ligand, and a linker which couples these two moieties together. In binding to its respective targets, the PROTAC recruits the HDAC to an E3 ligase, forming a ternary complex. Subsequent polyubiquitination of the HDAC enzyme occurs, which tags it for selective degradation via the proteasome. First reported in 2001 by Sakamoto et al., the PROTAC technology has received significant interest in recent years, particularly as a strategy to treat difficult-to-drug protein targets [39][40]. It has experienced a series of advancements in its design along the way, with the introduction of all small molecule-based PROTACs as well as incorporation of new E3 ligase ligands, affording a wide range of potent protein degraders [41][42][43][44]. In addition to demonstrating therapeutic effects, PROTACs have also proved important tools for biological discovery [45]. A key feature to the PROTAC mode of action is the ability to act in a catalytic manner. By incorporating a reversible target protein inhibitor into the design, a PROTAC can dissociate from the ternary complex following the polyubiquitination process, then bind to another target protein thereby enabling repeated cycles of induced protein degradation.
An HDAC enzyme degradation approach offers a number of potential advantages over inhibition regarding their use as future therapeutic agents. Firstly, PROTACs could enable reduced dosing and hence lower systemic drug exposure due to their catalytic mode of action and reduced occupancy level requirement. This may result in significantly reduced side effects in comparison to HDAC inhibitors, which require stoichiometric drug binding for modulation of protein function. Secondly, PROTACs offer the potential of a prolonged duration of action, as restoration of HDAC function will require re-synthesis of the enzyme. PROTACs are capable of multiple rounds of degradation, so following knockdown of HDAC levels, they can be envisaged to maintain low HDAC levels. Finally, alike the benefits of dual inhibitors over mono inhibitors, HDAC PROTACs provide another strategy for enhancing HDAC isoform or complex selectivity, as well as overcoming drug resistance.
In light of discoveries over the last couple of years, there are now a variety of HDAC-targeting PROTACs that have been developed, capable of degrading class-I, -II and -III HDACs (Figure 3). The first report of a HDAC-targeting PROTAC was in 2018 by Schiedel et al., who synthesised a SIRT2-selective degrader [46]. This was also the first evidence of the PROTAC technology being used to target an epigenetic eraser protein. As previously discussed, sirtuins (SIRT1-7) make up the class-III HDACs and belong to a different family to the class-I,-II and -IV HDACs, as they require a NAD+ cofactor for activity instead of Zn2+. The group had recently developed triazole-based SIRT2-selective inhibitors and already utilised these in synthesising biotinylated SIRT2 probes using Cu(I)-catalysed cycloaddition (“click”) chemistry [47]. The structural features from this work provided a rationale for the design of SIRT2 PROTACs, as the previously used alkynylated SIRT2 inhibitor was this time “clicked” to a cereblon E3 ligase ligand (thalidomide) equipped with a terminal azide handle. Docking of the anticipated ternary complex was used to guide choice of linker length, and the resulting PROTAC (29) demonstrated SIRT2 degradation of up to 90% at 5 μM in HeLa cells. Time course studies found that maximum effect was observed after 2 h of treatment.
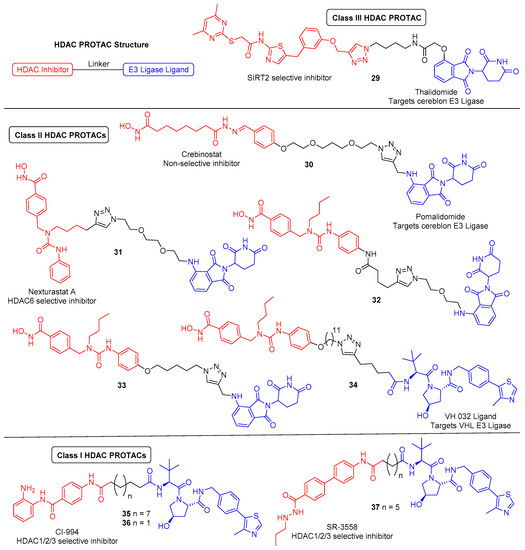
Figure 3. HDAC-targeting proteolysis-targeting chimeras (PROTACs). PROTAC components: HDAC inhibitor (red), small molecule E3 ligase ligand (blue), linker (black).
The first zinc-dependent HDAC to be successfully degraded via PROTACs was the class-II isoform HDAC6. Yang and co-workers conjugated the non-selective HDAC inhibitor crebinostat to the cereblon E3 ligase ligand pomalidomide via a polyethylene glycol (PEG) linker to afford 30 [48]. In multiple myeloma MM.1S cells, 30 achieved maximum effect and almost complete HDAC6 knockdown at 80 nM or higher concentrations, with no significant change in the protein levels of HDAC 1,2 or 4 under equivalent conditions. Therefore. despite incorporating a pan HDAC inhibitor, 30 demonstrated an impressive degree of isoform-selective degradation. The formation of a stable ternary complex has been shown to be a vital feature in order to achieve degradation of the target protein. This work suggests that 30 is only capable of forming a stable ternary complex with the cereblon E3 ligase and HDAC6, not any of the other HDACs, thereby providing the observed isoform selectivity. Although not degrading other HDAC enzymes, increased Ac-H3 levels were observed following treatment with 30, demonstrating that it still functioned as an inhibitor to other HDACs.
Following discovery of 30, there has since been a flurry of other HDAC6 degraders reported over the last couple of years by the Rao and Tang groups (PROTACs 31–34). All of these PROTACs have incorporated the HDAC6-selective inhibitor Nexturastat A and utilised “click” chemistry to conjugate the inhibitor to the E3 ligase ligand. A co-crystal structure of HDAC6 with Nexturastat A revealed that it binds via a y-shaped conformation, with the hydroxamic acid zinc binding group positioned inside the binding pocket for chelation to the zinc ion, and both the terminal benzene ring and aliphatic carbon chain protruding from the pocket, interacting with the isoform-specific hydrophobic surface rim of HDAC6 [49][50]. The latter surface exposed components thus served as ideal handles to build out from. In successive works, the Rao group investigated both such positions, tethering pomalidomide via PEG linkers to both the aliphatic chain and benzene ring of Nexturastat A, synthesising two sets of degraders [50][51]. Overall, they found that functionalising from both locations of Nexturastat A produced HDAC6-selective degraders. The most potent PROTAC in each case (31 and 32) displayed comparable activity and effectively induced selective HDAC6 degradation at 100 nM across different cell lines.
During this time, Wu et al. also “clicked” pomalidomide to the benzene ring of Nexturastat A, here using varying length alkyl linkers either side of the triazole ring to afford a range of potent and selective degraders [52]. The most potent PROTAC, 33, had a DC50 (degradation concentration to achieving half-maximal degradation) of 1.64 nM and could induce its maximal degradation (Dmax) of 86% at as low as 30 nM. Highlighted in this work is also the additional response caused by the incorporation of pomalidomide, which, along with thalidomide, makes up part of a class of drugs known as immunomodulatory imide drugs (IMiDs). These molecules, upon binding cereblon, can induce degradation of neo-substrates, most notably the Ikaros zinc finger (IKZF) proteins. Compound 33 was shown to induce IKZF1/3 degradation in addition to HDAC6 degradation, which resulted in promising antimyeloma activity due to synergistic effects. Although useful in this example, the side effects caused by IMiD containing PROTACs have been questioned to limit their utility. In a follow-up paper, the Tang group recently developed a related HDAC6-selective PROTAC, 34, now incorporating a different E3 ligase ligand which targets the Von Hippel–Lindau (VHL) E3 ubiquitin ligase [53]. Interestingly, a much longer linker length was required for efficient levels of degradation in comparison to the cereblon-based degraders. Compound 34 maintained potent HDAC degradation (DC50 = 7.1 nM, Dmax = 90%) and, as expected, caused no induced degradation of IKZF1/3 proteins.
Unlike the class-II, -III and -IV HDACs, the class-I HDACs are nucleus localised and exist as part of much larger multiprotein corepressor complexes in vivo. Recently, we reported the first example of a class-I HDAC-targeting PROTAC, which demonstrated degradation of HDACs 1,2 and 3 [54]. The class-I o-aminoanilide HDAC inhibitor CI-994 was conjugated to the VHL ligand via a twelve-carbon alkyl linker to synthesise PROTAC 35. After 24 h treatment in HCT116 human colon cancer cells, 35 achieved almost complete degradation of HDACs 1 and 2 at 10 µM, whilst HDAC3 levels were also decreased. At least 50% degradation was observed for HDACs 1,2 and 3 at 1 µM. Linker lengths incorporating six-carbon linkers with either VHL (36) or thalidomide failed to degrade class-I HDACs in cells despite retaining sub micromolar HDAC inhibition in vitro. Using the HDAC2 crystal structure with a directly analogous o-aminoanilide inhibitor bound (PDB 4LY1) in the HDAC active site, and the pVHL:EloB:EloC complex with the VHL E3-ligase ligand bound (PDB 4W9H), these proteins were modelled by tethering their respective ligands together via a twelve carbon linker and six carbon linker, recreating 35 and 36 from our study (Figure 4). It can be seen with the twelve-carbon linker, 35, VHL and HDAC2 are brought into close proximity with no direct steric clash or protein–protein overlap, unlike the six-carbon linker, 36, suggesting that if 36 is cell permeable, it may be unable to form the necessary ternary complex required for polyubiquitination and degradation.
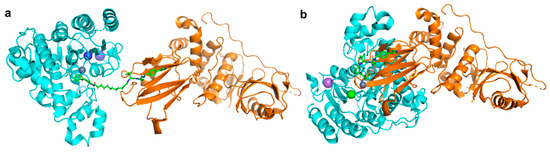
Following the discovery of 35, Xiao et al. reported the HDAC3 specific PROTAC 37, which like 35 is a VHL-recruiting degrader and utilises an alkyl linker in its design [57]. The incorporated HDAC inhibitor SR-3558 was previously identified by the group as being class-I selective and features a novel benzoylhydrazide ZBG [58]. In MDA-MB-468 cells, 37 selectively induced HDAC3 degradation with a DC50 concentration of 42 nM following a 14 h treatment, with little to no change in HDAC 1,2 or 6 protein levels. Washout assays revealed the impact of 37 on HDAC3 protein levels was long-lasting and reversible, requiring over 12 h to restore to pre-treatment levels. With an enhanced selectivity profile observed for 37 compared to the parent inhibitor, future research in this area may lead to the development of HDAC complex selective or other class-I HDAC isoform-selective PROTACs.
3. Conclusions and Future Outlook
The FDA approval of five HDAC inhibitors has firmly established the importance of HDACs as a therapeutically relevant target and paved the way for the development of new inhibitor designs to achieve improved efficacy and selectivity profiles. HDACs are associated with various multifactorial diseases, hence, combining HDAC inhibition with that of another disease-related target has emerged as a valuable approach to deliver a more directed and sustained treatment over single-targeting agents. This polypharmacological treatment has been successfully achieved using bifunctional HDAC therapeutics—single molecule, dual-targeting agents comprising a HDAC inhibitor conjugated to another specificity-targeting moiety. In many cases, dual HDAC inhibitors have been developed following observed synergy between HDAC inhibitors in combinatorial therapy with other drugs. The HDAC inhibitor pharmacophore can tolerate extensive cap modification and so is very amenable to the synthesis of such hybrids. Already, an extensive list of targets has now been successfully inhibited via dual HDAC inhibitors, complete with impressive potency and high levels of HDAC isoform or class selectivity. This list is set to expand in the years ahead.
The class-I HDACs exist as part of multi-protein complexes in vivo, presenting another factor to consider for inhibition along with the high homology present between the isoforms. Dual HDAC inhibitors offer the potential to target the HDAC and one of its protein partners within a specific complex, providing a novel strategy to achieve complex selectivity. This approach has already been achieved in targeting the CoREST complex, concurrently inhibiting both HDAC and LSD1, but has the potential to be extended to other HDAC co-repressor complexes.
PROTACs are an interesting deviation for bifunctional HDAC modulators, offering an alternative strategy for achieving enhanced selectivity over inhibition alone. Whilst still in its infancy, recent findings are delivering highly selective and potent HDAC degraders, thus sparking a new area for discovery of potential future therapeutics. Development of complex-selective HDAC PROTACs would be an exciting prospect as a new means to treat complex-specific diseases.
References
- Mai, A.; Massa, S.; Rotili, D.; Cerbara, I.; Valente, S.; Pezzi, R.; Simeoni, S.; Ragno, R. Histone deacetylation in epigenetics: An attractive target for anticancer therapy. Med. Res. Rev. 2005, 25, 261–309.
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705.
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840.
- Zhao, S.; Xu, W.; Jiang, W.; Yu, W.; Lin, Y.; Zhang, T.; Yao, J.; Zhou, L.; Zeng, Y.; Li, H.; et al. Regulation of cellular metabolism by protein lysine acetylation. Science 2010, 327, 1000–1004.
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552.
- Gregoretti, I.; Lee, Y.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31.
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39.
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Disc. 2014, 13, 673–691.
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432.
- Aldana-Masangkay, G.I.; Sakamoto, K.M. The role of HDAC6 in cancer. J. Biomed. Biotechnol. 2011, 2011, 875824.
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2008, 277, 8–21.
- Kilgore, M.; Miller, C.A.; Fass, D.M.; Hennig, K.M.; Haggarty, S.J.; Sweatt, J.D.; Rumbaugh, G. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 2010, 35, 870–880.
- Thomas, E.A.; Coppola, G.; Desplats, P.A.; Tang, B.; Soragni, E.; Burnett, R.; Gao, F.; Fitzgerald, K.M.; Borok, J.F.; Herman, D.; et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc. Natl. Acad. Sci. USA 2008, 105, 15564–15569.
- Sumner, C.J.; Huynh, T.N.; Markowitz, J.A.; Perhac, J.S.; Hill, B.; Coovert, D.D.; Schussler, K.; Chen, X.; Jarecki, J.; Burghes, A.H.M.; et al. Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann. Neurol. 2003, 54, 647–654.
- Jenssen, K.; Burnett, R.; Soragni, E.; Herman, D.; Perlman, S.L.; Gottesfeld, J.M. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat. Chem. Biol. 2006, 2, 551–558.
- Chen, K.; Zhang, X.; Wu, Y.; Wiest, O. Inhibition and mechanism of HDAC8 revisited. J. Am. Chem. Soc. 2014, 136, 11636–11643.
- Gantt, S.M.L.; Decroos, C.; Lee, M.S.; Gullett, L.E.; Bowman, C.M.; Christianson, D.W.; Fierke, C.A. General base–general acid catalysis in human histone Deacetylase 8. Biochemistry 2016, 55, 820–832.
- Roche, J.; Bertrand, P. Inside HDACs with more selective HDAC inhibitors. Eur. J. Med. Chem. 2016, 121, 451–483.
- Dokmanovic, M.; Marks, P.A. Prospects: Histone deacetylase inhibitors. J. Cell. Biochem. 2005, 96, 293–304.
- Lane, A.A.; Chabner, B.A. Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 2009, 27, 5459–5468.
- McKinsey, T.A. Isoform-selective HDAC inhibitors: Closing in on translational medicine for the heart. J. Mol. Cell. Cardiol. 2010, 51, 491–496.
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA Approval summary: Vorinostat for treatment of advanced primary cutaneous T-Cell lymphoma. Oncologist 2007, 12, 1247–1252.
- VanderMolen, K.M.; McCulloch, W.; Pearce, C.J.; Oberlies, N.H. Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): A natural product recently approved for cutaneous T-cell lymphoma. J. Antibiot. 2011, 64, 525–531.
- Laubach, J.P.; Moreau, P.; San-Miguel, J.F.; Richardson, P.G. Panobinostat for the treatment of multiple myeloma. Clin. Cancer Res. 2015, 21, 4767–4773.
- Poole, R. Belinostat: First global approval. Drugs 2014, 74, 1543–1554.
- Lu, X.; Ning, Z.; Li, Z.; Cao, H.; Wang, X. Development of chidamide for peripheral T-cell lymphoma, the first orphan drug approved in China. Intractable Rare Dis. Res. 2016, 5, 185–191.
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty years of HDAC inhibitors: 2020 insight and hindsight. J. Med. Chem. 2020.
- Duvic, M.; Talpur, R.; Ni, X.; Zhang, C.; Hazarika, P.; Kelly, C.; Chiao, J.H.; Reilly, J.F.; Ricker, J.L.; Richon, V.M.; et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007, 109, 31–39.
- Vansteenkiste, J.; Van Cutsem, E.; Dumez, H.; Chen, C.; Ricker, J.; Randolph, S.; Schöffski, P. Early phase II trial of oral vorinostat in relapsed or refractory breast, colorectal, or non-small cell lung cancer. Investig. New Drugs 2008, 26, 483–488.
- Schneider, B.; Kalemkerian, G.; Bradley, D.; Smith, D.; Egorin, M.; Daignault, S.; Dunn, R.; Hussain, M. Phase I study of vorinostat (suberoylanilide hydroxamic acid, NSC 701852) in combination with docetaxel in patients with advanced and relapsed solid malignancies. Investig. New Drugs 2012, 30, 249–257.
- Shah, M.H.; Binkley, P.; Chan, K.; Xiao, J.; Arbogast, D.; Collamore, M.; Farra, Y.; Young, D.; Grever, M. Cardiotoxicity of histone deacetylase inhibitor depsipeptide in patients with metastatic neuroendocrine tumors. Clin. Cancer Res. 2006, 12, 3997–4003.
- DeAngelo, D.J.; Mesa, R.A.; Fiskus, W.; Tefferi, A.; Paley, C.; Wadleigh, M.; Ritchie, E.K.; Snyder, D.S.; Begna, K.; Ganguly, S.; et al. Phase II trial of panobinostat, an oral pan-deacetylase inhibitor in patients with primary myelofibrosis, post-essential thrombocythaemia, and post-polycythaemia vera myelofibrosis. Br. J. Haematol. 2013, 162, 326–335.
- Whitehead, R.P.; Rankin, C.; Hoff, P.M.G.; Gold, P.J.; Billingsley, K.G.; Chapman, R.A.; Wong, L.; Ward, J.H.; Blanke, C.D. Phase II trial of depsipeptide (NSC-630176) in previously treated colorectal cancer patients with advanced disease: A southwest oncology group study (S0336). Investig. New Drugs 2009, 27, 469–475.
- Luu, T.H.; Morgan, R.J.; Leong, L.; Lim, D.; McNamara, M.; Portnow, J.; Frankel, P.; Smith, D.D.; Doroshow, J.H.; Gandara, D.R.; et al. A Phase II Trial of vorinostat (suberoylanilide hydroxamic acid) in metastatic breast cancer: A California cancer consortium study. Clin. Cancer Res. 2008, 14, 7138–7142.
- Traynor, A.M.; Dubey, S.; Eickhoff, J.C.; Kolesar, J.M.; Schell, K.; Huie, M.S.; Groteluschen, D.L.; Marcotte, S.M.; Hallahan, C.M.; Weeks, H.R.; et al. Vorinostat (NSC# 701852) in patients with relapsed non-small cell lung cancer: A wisconsin oncology network phase II study. J. Thorac. Oncol. 2009, 4, 522–526.
- Woyach, J.A.; Kloos, R.T.; Ringel, M.D.; Arbogast, D.; Collamore, M.; Zwiebel, J.A.; Grever, M.; Villalona-Calero, M.; Shah, M.H. Lack of therapeutic effect of the histone deacetylase inhibitor vorinostat in patients with metastatic radioiodine-refractory thyroid carcinoma. J. Clin. Endocrinol. Metab. 2009, 94, 164–170.
- Stadler, W.M.; Margolin, K.; Ferber, S.; McCulloch, W.; Thompson, J.A. A phase II study of depsipeptide in refractory metastatic renal cell cancer. Clin. Genitourin. Cancer 2006, 5, 57–60.
- Hainsworth, J.D.; Infante, J.R.; Spigel, D.R.; Arrowsmith, E.R.; Boccia, R.V.; Burris, H.A. A phase II trial of panobinostat, a histone deacetylase inhibitor, in the treatment of patients with refractory metastatic renal cell carcinoma. Cancer Investig. 2011, 29, 451–455.
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559.
- Nalawansha, D.A.; Crews, C.M. PROTACs: An emerging therapeutic modality in precision medicine. Cell Chem. Biol. 2020, 27, 998–1014.
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med. Chem. Lett. 2008, 18, 5904–5908.
- Bondeson, D.P.; Mares, A.; Smith, I.E.D.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617.
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.; Crew, A.; Coleman, K.; et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 2015, 22, 755–763.
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381.
- Burslem, G.M.; Crews, C.M. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell 2020, 181, 102–114.
- Schiedel, M.; Herp, D.; Hammelmann, S.; Swyter, S.; Lehotzky, A.; Robaa, D.; Oláh, J.; Ovádi, J.; Sippl, W.; Jung, M. Chemically induced degradation of sirtuin 2 (Sirt2) by a proteolysis targeting chimera (PROTAC) based on sirtuin rearranging ligands (SirReals). J. Med. Chem. 2018, 61, 482–491.
- Schiedel, M.; Rumpf, T.; Karaman, B.; Lehotzky, A.; Gerhardt, S.; Ovádi, J.; Sippl, W.; Einsle, O.; Jung, M. Structure-based development of an affinity probe for Sirtuin 2. Angew. Chem. Int. Ed. 2016, 55, 2252–2256.
- Yang, K.; Song, Y.; Xie, H.; Wu, Y.; Wu, H.; Leisten, E.D.; Tang, W. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorg. Med. Chem. Lett. 2018, 28, 2493–2497.
- Miyake, Y.; Keusch, J.J.; Wang, L.; Saito, M.; Hess, D.; Wang, X.; Melancon, B.J.; Helquist, P.; Gut, H.; Matthias, P. Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat. Chem. Biol. 2016, 12, 748–754.
- Yang, H.; Lv, W.; He, M.; Deng, H.; Li, H.; Wu, W.; Rao, Y. Plasticity in designing PROTACs for selective and potent degradation of HDAC6. Chem. Commun. 2019, 55, 14848–14851.
- An, Z.; Lv, W.; Su, S.; Wu, W.; Rao, Y. Developing potent PROTACs tools for selective degradation of HDAC6 protein. Protein Cell 2019, 10, 606–609.
- Wu, H.; Yang, K.; Zhang, Z.; Leisten, E.D.; Li, Z.; Xie, H.; Liu, J.; Smith, K.A.; Novakova, Z.; Barinka, C.; et al. Development of multifunctional histone deacetylase 6 degraders with potent antimyeloma activity. J. Med. Chem. 2019, 62, 7042–7057.
- Yang, K.; Wu, H.; Zhang, Z.; Leisten, E.D.; Nie, X.; Liu, B.; Wen, Z.; Zhang, J.; Cunningham, M.D.; Tang, W. Development of selective histone deacetylase 6 (HDAC6) degraders recruiting von hippel-lindau (VHL) E3 ubiquitin ligase. ACS Med. Chem. Lett. 2020, 11, 575–581.
- Smalley, J.P.; Adams, G.E.; Millard, C.J.; Song, Y.; Norris, J.K.S.; Schwabe, J.W.R.; Cowley, S.M.; Hodgkinson, J.T. PROTAC-mediated degradation of class I histone deacetylase enzymes in corepressor complexes. Chem. Commun. 2020, 56, 4476–4479.
- Lauffer, B.E.L.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; Flicke, B.; Ritscher, A.; Fedorowicz, G.; Vallero, R.; et al. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 2013, 288, 26926–26943.
- Galdeano, C.; Gadd, M.S.; Soares, P.; Scaffidi, S.; Van Molle, I.; Birced, I.; Hewitt, S.; Dias, D.M.; Ciulli, A. Structure-guided design and optimization of small molecules targeting the protein-protein interaction between the von Hippel–Lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. J. Med. Chem. 2014, 57, 8657–8663.
- Xiao, Y.; Wang, J.; Zhao, L.Y.; Chen, X.; Zheng, G.; Zhang, X.; Liao, D. Discovery of histone deacetylase 3 (HDAC3)-specific PROTACs. Chem. Commun. 2020, 56, 9866–9869.
- Wang, Y.; Stowe, R.; Pinello, C.; Tian, G.; Madoux, F.; Li, D.; Zhao, L.; Li, J.; Wang, Y.; Wang, Y.; et al. Identification of histone deacetylase inhibitors with benzoylhydrazide scaffold that selectively inhibit class I histone deacetylases. Chem. Biol. 2015, 22, 273–284.


