 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Francesca Bufalieri | + 2859 word(s) | 2859 | 2020-09-30 08:08:18 | | | |
| 2 | Bruce Ren | Meta information modification | 2859 | 2020-10-09 03:23:01 | | | | |
| 3 | Francesca Bufalieri | Meta information modification | 2859 | 2021-02-04 13:30:56 | | |
Video Upload Options
The Hedgehog (HH) pathway governs cell proliferation and patterning during embryonic development and is involved in regeneration, homeostasis and stem cell maintenance in adult tissues. The activity of this signaling is finely modulated at multiple levels and its dysregulation contributes to the onset of several human cancers. Ubiquitylation is a coordinated post-translational modification that controls a wide range of cellular functions and signaling transduction pathways. It is mediated by a sequential enzymatic network, in which ubiquitin ligase (E3) and deubiquitylase (DUBs) proteins are the main actors. The dynamic balance of the activity of these enzymes dictates the abundance and the fate of cellular proteins, thus affecting both physiological and pathological processes. Several E3 ligases regulating the stability and activity of the key components of the HH pathway have been identified. Further, DUBs have emerged as novel players in HH signaling transduction, resulting as attractive and promising drug targets.
1. The HH Signaling Pathway and Tumorigenesis: An Overview
The HH pathway is a mitogen and morphogen signaling, conserved from Drosophila to mammals. It plays a crucial role in organogenesis and central nervous system (CNS) development [1][2][3][4]. In post-embryonic stages, HH signaling regulates tissue homeostasis and repair, modulating the specification of the adult stem cells [5][6]. Several studies have highlighted similarities and divergences between Drosophila and mammals HH signal transduction (Figure 1A,1B). Both in flies and in vertebrates the HH pathway activation is finely orchestrated by two membrane receptors: the multi-pass transmembrane protein Patched (Ptc/PTCH) and the heptahelical transmembrane co-receptor Smoothened (Smo/SMO). In Drosophila, in absence of the Hh ligand, Ptc keeps off the signaling by directly affecting Smo activity and preventing its accumulation on the plasma membrane. In this state, Costal-2 (Cos2; Costa-FlyBase), a kinesin family protein, Fused (Fu), a serine-threonine kinase and the Suppressor of fused [Su(fu)] inhibit the bifunctional transcription factor Cubitus interrupts (Ci), endowed of both repressor and activator domains. The full-length Ci protein is proteolytically processed by the Skp1-Cullin1-Slimb (SCFSlimb) ubiquitin ligase complex, in a truncated form (CiR) that acts as transcriptional repressor of Hh target genes when translocated into the nucleus (Figure 1A) [3][4].
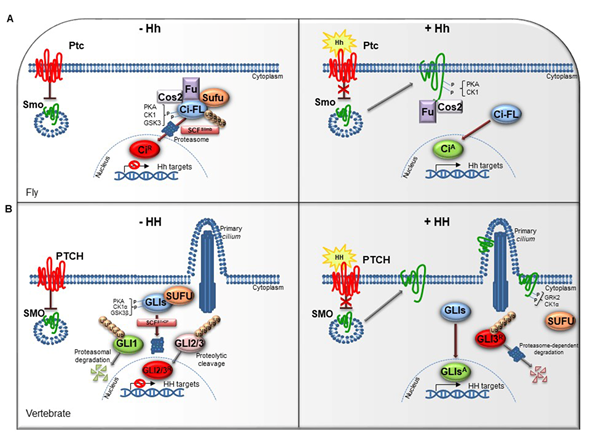
Figure 1. The Hedgehog signaling pathway. (A) The Hedgehog signaling pathway in fly. In absence of Hh, Ptc inhibits the localization of Smo on cell membrane. In the cytoplasm, Cos2, Fu and Sufu assemble in complex with Ci-FL protein, favoring its phosphorylation by PKA, CK1, and GSK3. This event induces the Ci-FL ubiquitylation by SCFSlimb E3 ligase thus leading both to proteasome degradation and cleavage into truncated repressor form (CiR). CiR blocks the transcription of Hh target genes. On the contrary, in the presence of Hh ligand, Ptc releases the inhibitory effect exerted on Smo which is activated by PKA and CK1 phosphorylation on the C-terminal domain, and then bound by Cos2 and Fu. These processes culminate in the Ci activation, promoting Hh transcription. (B) The Hedgehog signaling pathway in vertebrates. When the pathway is turned off, PTCH prevents the accumulation of SMO in the primary cilium. SUFU restrains GLI transcription factors in the cytoplasm where PKA, CK1α, and GSK3β kinases promote their phosphorylation. This process attracts the SCFβTrCP E3 ligase that determines the processing of GLI2 and GLI3 (GLI2/3R) in their repressor forms and the proteasome-mediated degradation of GLI1. In presence of HH ligand, PTCH inhibition is relieved. SMO is accumulated in the primary cilium and activated by GRK2 and CK1α phosphorylation. GLI activator forms (GLIsA) translocate into the nucleus and induce the transcription of HH target genes.
In mammals, three ligands belonging to the HH family are secreted: Desert hedgehog (DHH), Indian hedgehog (IHH) and Sonic hedgehog (SHH). The proteins, encoded by three paralogous mammalian genes, share high similarity in the affinity with HH-binding proteins. SHH is mostly expressed in brain cells and implicated in central nervous system (CNS) development, while IHH modulates chondrogenesis, and DHH regulates spermatogenesis and nerve-Schwann cell interactions [7][8][9].
A peculiar characteristic of HH signal transduction is the role of the primary cilium. This organelle is a microtubule-based protrusion of the cell membrane that coordinates protein trafficking events, recruits and stabilizes a regulative dynamic network among the core of HH components [10].
The complexity of HH signaling in vertebrates is also provided by the GLI zinc-finger transcription factors, the final effectors of the pathway (Figure 1B). Three GLI members have been identified, GLI1, GLI2 and GLI3: GLI1 acts exclusively as activator, instead GLI2 and GLI3, which have an N-terminal repressor domain, can work either as repressors or activators [11]. The balance between activator and repressor forms is widely ruled by SUFU, an essential negative regulator that controls HH signaling through its direct interaction with GLI factors [11][12].
When the HH pathway is off, phosphorylated GLI1 is recognized by the Skp1-Cullin1-βTrCP (SCFβTrCP) ubiquitin ligase complex and degraded by proteasome system, whereas GLI2 and GLI3 undergo a proteolytic process that converts them into cleaved transcriptional repressor forms. Otherwise, the binding of mature HH ligand to PTCH receptor releases the inhibition exerted on SMO, resulting in its activation and translocation into the cilium. These events lead to the nuclear localization of GLI activator forms where they induce the expression of HH-target genes, which include GLI1 itself, thus triggering a positive feedback loop that amplifies the signal [13][14].
The HH pathway output is tightly regulated at multiple levels by different post-translational modifications, such as phosphorylation and ubiquitylation [15][16][17]. The pattern of GLI phosphorylation triggered by the protein kinase A (PKA), the casein kinase 1 (CK1α) and the glycogen synthase kinase 3 (GSK3β) establishes multiple states of GLI activity and ultimately influences the HH transcriptional program [18]. The sequential phosphorylation of GLI proteins leads to the recruitment of the SCFβTrCP, thus promoting GLI ubiquitylation and proteasome-mediated processing, as also described for its homolog Ci in Drosophila [19].
The ubiquitin-mediated processes of GLI factors are also triggered by other E3 ligases, such as the RING Cullin3-HIB/Roadkill/SPOP complex, the acetyltransferase/E3 ligase PCAF (P300/CBP-associated factor), and the HECT E3 ligase Itch. Importantly, Itch controls HH signaling by distinct routes: it mediates regulatory events on SUFU and proteasome degradation of GLI1 and PTCH1 by the interaction with the adaptor proteins β-arrestin2 and Numb, respectively [20][8][9][21][22][23][24][25].
In the last years, post-translational modifications have also been described to control SMO activity. As GLIs, SMO is regulated, in response to HH stimuli, by PKA/CK1-mediated phosphorylation in Drosophila and GRK2/CK1α in mammals, and downregulated by ubiquitin-mediated endocytosis and ubiquitin-dependent lysosome or proteasome degradation [26]. In Drosophila, Smo ubiquitylation and trafficking on cell surface is regulated by the HECT E3 ligases Smurf and Herc4, and the E3 ligase complex formed by Cullin4 and DNA-damage-binding protein 1 (DDB1), recruited by Smo through the β subunit of trimeric G protein (Gβ) [27][28]. Moreover, in mammals HERC4 has been described as tumor suppressor in non-small cell lung cancer (NSCLC) able to control SMO protein stability [29].
Given the essential role of HH signaling for a proper development, mutations in its key players cause congenital malformations [30]. An uncontrolled and permanent activity of the HH pathway is also associated to various human cancers such as basal cell carcinoma (BCC), medulloblastoma (MB), gliomas, pancreatic, colorectal, prostate, lung, and breast cancers (Figure 2) [31][32][33]. Indeed, aberrant HH activation involves and triggers pro-tumorigenic events, such as proliferation, survival, angiogenesis, migration and epithelial-mesenchymal transition (EMT) [34], thus affecting every step of carcinogenesis, from early development to metastatic progression [31][32].
Hyperactivation of HH signaling can occurs through either ligand-independent or ligand-dependent mechanisms. Tumorigenesis is ligand-independent when the pathway is constitutively activated in the absence of ligand via mutations in HH signaling components. Loss-of-function mutations in PTCH or SUFU or gain-of-function mutations in SMO, as well as GLI1 overexpression or GLI2 amplification have been identified in BCC, a common human skin cancer, and in MB, a highly malignant pediatric brain tumor [35][36][37][38][39]. Depending on the type of HH ligand release, two mechanisms of ligand-dependent pathway hyperactivation have been described in cancers, generating a tumor-stromal crosstalk [40]. Ligand-dependent autocrine/juxtacrine secretion occurs when the HH ligand is profusely released and caught by the same tumor cells, thus activating the pathway. Tumors that arise from this condition may display HH ligand overexpression or high levels of PTCH1 and GLI1 [41][42][43]. Alternatively, a paracrine secretion of HH ligand by tumor cells can induce the activation of the HH pathway in stromal cells of tumor microenvironment. As consequence, the stroma secretes paracrine growth signals to induce tumor growth [44]. For instance, in prostate cancer specimens, the expression of HH was detected in the tumor epithelium, while GLI1 expression was found in the tumor stroma cells, suggesting their paracrine crosstalk [45]. Moreover, this mechanism of HH signaling activation can work in a reverse paracrine manner in which cancer cells take the HH ligand released by stromal cells. For example, HH ligand released by bone marrow, nodal and splenic stroma can activate the HH pathway and maintain the survival of B and plasma cells in hematological malignancies [46]. Interestingly, HH-producing microenvironment is required for GLI activation in gliomas [47].
Figure 2. Schematic representation of HH-related tumors. The hyperactivation of HH signaling is involved in the tumorigenesis of several human malignancies here reported.
Of note, HH signaling also regulates the expression of the stemness genes Nanog and Oct4, thus participating in the formation or maintenance of cancer stem cells (CSCs) responsible of tumor initiation, relapse and drug resistance [48][49][50]. For all these reasons, the HH pathway is emerged as an attractive druggable target for anti-cancer therapy. A various number of SMO antagonists, able to block the pathway at upstream level, have been identified and patented. Some of them, vismodegib and sonidegib, and recently glasdegib, have been approved by the Food and Drug Administration (FDA) for the treatment of BCC and Acute Myeloid Leukemia (AML), respectively . Many others, such as GANT61 and GlaB, have been designed targeting GLI1, the downstream effector of HH signaling, and have shown efficacy in preclinical study [51][52]. The major issue in employment of HH-inhibitors is the recurrence of drug-resistance mutations or alternative mechanisms of activation. Consequently, multi-target therapy is emerging as a promising strategy for the treatment of HH-dependent cancers. The best approach envisioned so far is the development of further inhibitors, or the identification of additional regulators of the HH pathway that could be targeted in tumorigenesis.
2. Ubiquitylation Process
Ubiquitylation dictates the fate and function of most cellular proteins increasing the complexity of the proteome. This modification is a dynamic and tightly regulated post-translational event with many distinct outcomes affecting protein stability, localization, interactions, and activity.
Ubiquitin (Ub) is a small globular protein consisting of 76 amino acids encoded in mammals by four different genes (UBB, UBC, RPS27, and UBA52) that ensure high cellular Ub levels [53]. Ubiquitylation is a multi-step process orchestrated by an enzymatic cascade that relies on Ub and three different enzymes: Ub-activating (E1), Ub-conjugating (E2), and Ub-ligating (E3) [54]. During the catalytic reactions, Ub is activated in an ATP-dependent way by an E1 enzyme, subsequently transferred to the active cysteine (Cys) residue of an E2 enzyme via a trans-(thio) esterification reaction, and finally attached with an isopeptide bond to a substrate by an E3 enzyme (Figure 3A). In humans, two E1s, around 30 E2s and over 600 E3s have been identified [55][56]. The latter are the major determinants and provide specificity for substrate recognition. Based on their functional domains and on the mechanism of catalysis, E3s are divided into three main families: the Really Interesting New Gene (RING), the Homologous to the E6-associated protein Carboxyl-Terminus (HECT) types, and RING-between-RING (RBR), which can be considered a RING-HECT hybrid [57][58]. Each class of E3 ligases can create Ub linkages of different length and architecture. The transfer of the Ub moiety to substrate occurs through the formation of the covalent bond between α-carboxyl group of the terminal glycine (Gly) residue of Ub and, commonly, ε-amino group of an internal lysine (Lys) residue of the substrate. Of note, for a subset of substrates the attachment of Ub may interest their N-terminal residue, a process known as N-terminal ubiquitylation [59], or serine and threonine residues, further expanding the complexity and the biological relevance of this process. In this regard, Ub modifications of a target protein occur in various forms: attachment of a single Ub moiety on a single substrate residue (monoubiquitylation), a single Ub on multiple residues (multi-ubiquitylation), or additional Ub molecules to initial Ub yielding an ubiquitin chain (poly-ubiquitylation). Typically, mono- and multiubiquitylation regulate endocytosis, signal transduction, DNA repair, and often result in changes in the cellular localization and protein activity[60][62]. By contrast, polyubiquitylation is the most abundant modification that controls protein homeostasis. Indeed, the polyubiquitylated target substrates are recognized by the 26S proteasome, a multiprotein complex, that degrades the proteins into small peptides and releases the Ub for cyclic utilization [63]. Besides regulating protein degradation, polyubiquitylation brings different functional consequences depending on Ub chain linkage-type [64]. Ub has seven Lys residues (K6, K11, K27, K29, K33, K48, and K63) that may serve as polyubiquitylation points. Depending upon the Lys used, length of the chains and linkage type, distinctive forms of Ub chains may be achieved to drive the fate of target proteins [65]. Lys48-linkage targets protein for proteasome-dependent degradation, whereas Lys63-linkage is associated to regulative processes, including trafficking, protein localization, protein-protein interaction; the biological significance of other Ub modifications is still largely unclear [66]. Further complexity is provided by Ub modifications (i.e., phosphorylation, acetylation, sumoylation) and by the linkage of Ub to other Ub-like proteins (i.e., NEDD8, SUMO), creating a multitude of distinct signals. The combination of all these parameters has been referred as the “Ub code” [65]. The Ub code governs the fate of the targeted substrates by modulating their interactions with many other proteins that incorporate Ub-binding domains and determine their accessibility to deubiquitylating enzymes (DUBs), a family of protease conserved from yeast to humans [67].
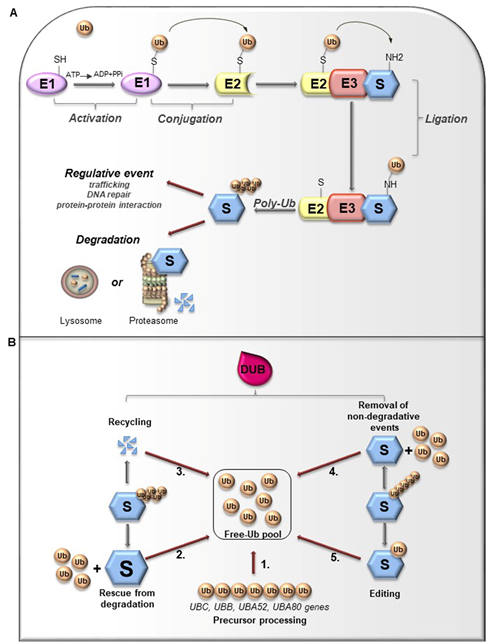
Figure 3. (A). Ubiquitylation processes. Ubiquitylation is a multi-step process that involves three enzymes: E1 (Ub-activating enzyme), E2 (Ub-conjugating enzyme) and E3 (Ub-ligase). Initially, Ub is linked to E1 through a high energy thioester bond. After, Ub activated by E1 is conjugated to a sulfhydryl group on E2 enzyme. Finally, E3 ligase specifically catalyzes the transfer of Ub from E2 to a Lys residue on a substrate protein. The formation of a poly-ubiquitin (poly-Ub) chain can lead the substrate toward a degradative or regulative pathway. (B). Deubiquitylation and DUBs function. Ubiquitylation can be reversed by deubiquitylating enzymes (DUBs) that hydrolyze the isopeptide or peptide bond, leading to Ub deconjugation from the ubiquitylated protein. DUBs have many functions. 1. Precursor processing: Ub is encoded by four genes and translated as a linear fusion protein consisting of multiple Ub copies, which require the cleavage by DUBs in order to generate free single Ub; 2. Rescue from degradation: DUBs can rescue protein from proteasomal or lysosome degradation; 3. Recycling: DUBs maintain Ub homeostasis preventing its degradation following substrate proteolysis; 4. Removal of non-degradative events: DUBs can remove Ub chains from substrates that are not committed to degradation; 5. Editing: DUBs can also affect the fate of ubiquitylated substrates by cleaving inter-Ub chains (switching from degradative to non-degradative ubiquitylation).
3. Deubiquitylating Enzymes: Functions and Classification
Like other important post-translational modifications, ubiquitylation is a dynamic and reversible process counteracted by DUBs activity [65]. DUBs are proteases that hydrolyze isopeptide or peptide bond removing Ub conjugates from substrates and disassembling anchored Ub chains (Figure 3B) [65][68]. DUBs may remove Ub moieties from the distal end or through the cleavage within chains in two distinct ways: i) via direct interaction with specific substrates; ii) through selective recognition for particular Ub chain architecture. Both chain length and linkage type may drive the choice of the target proteins. Importantly, linkage selectivity may occur within the catalytic domain or through the cooperation with Ub-binding domains within DUBs or their interaction partners [68].
Given their crucial role in opposing E3 ligases function, DUBs control protein homeostasis and activities, and are implicated in the regulation of various physiological and pathological processes, such as development, metabolism, immune response and tumorigenesis.
Currently, 99 cellular DUBs have been identified and are classified into six main families depending on distinct catalytic domains: the largest group ubiquitin-specific proteases (USPs), ubiquitin C-terminal hydrolases (UCHs), ovarian tumor proteases (OTUs), JAD/PAD/MPN-domain containing metalloenzymes (JAMMs), Machado-Joseph disease domain proteases (MJDs or Josephins) and motif interacting with Ub-containing novel DUB family (MINDYs) [69][70]. Unlike of the JAMM family, classified as a zinc-dependent metalloproteinase, the other DUBs classes are cysteine proteases. Available data indicate that each family may display linkage or substrate preferences. For instance, OTU family exhibits linkage type specificity, whereas USP group members show differences in catalytic rate constants [71][72]. Studies aimed at defining the abundance of individual DUBs suggest that those with constitutive functions show high copy number, while DUBs with peculiar roles are the rarer forms [70]Different approaches used to determine the intracellular localization of the DUBs allowed highlighting that subsets of these proteases show particular association with subcellular compartments. Although many DUBs are nuclear, several USP members localize to defined structure including plasma membrane, microtubules, endosome, and endoplasmic reticulum (ER) [73].
To date, a growing body of evidence indicated that DUBs can act as oncogenes or tumor suppressors emerging as a promising class of therapeutic targets. For these reasons, many efforts are devoted to the development of highly selective DUBs inhibitors for anti-cancer therapies.
References
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087, doi:10.1101/gad.938601.
- Briscoe, J.; Thérond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429, doi:10.1038/nrm3598.
- Lee, R.T.; Zhao, Z.; Ingham, P.W. Hedgehog signalling. Development 2016, 143, 367–372, doi:10.1242/dev.120154.
- Huangfu, D.; Anderson, K.V. Signaling from Smo to Ci/Gli: Conservation and divergence of Hedgehog pathways from Drosophila to vertebrates. Development 2006, 133, 3–14, doi:10.1242/dev.02169.
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and mechanisms. Genes Dev. 2008, 22, 2454–2472, doi:10.1101/gad.1693608.
- Petrova, R.; Joyner, A.L. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development 2014, 141, 3445–3457, doi:10.1242/dev.083691.
- Pathi, S.; Pagan-Westphal, S.; Baker, D.P.; Garber, E.A.; Rayhorn, P.; Bumcrot, D.; Tabin, C.J.; Blake Pepinsky, R.; Williams, K.P. Comparative biological responses to human Sonic, Indian, and Desert hedgehog. Mech. Dev. 2001, 106, 107–117, doi:10.1016/s0925-4773(01)00427-0.
- Jiang, J. Regulation of Hh/Gli signaling by dual ubiquitin pathways. Cell Cycle 2006, 5, 2457–2463, doi:10.4161/cc.5.21.3406.
- Wang, C.; Pan, Y.; Wang, B. Suppressor of fused and Spop regulate the stability, processing and function of Gli2 and Gli3 full-length activators but not their repressors. Development 2010, 137, 2001–2009, doi:10.1242/dev.052126.
- Kong, J.H.; Siebold, C.; Rohatgi, R. Biochemical mechanisms of vertebrate hedgehog signaling. Development 2019, 146, doi:10.1242/dev.166892.
- Hui, C.C.; Angers, S. Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 513–537, doi:10.1146/annurev-cellbio-092910-154048.
- Infante, P.; Faedda, R.; Bernardi, F.; Bufalieri, F.; Lospinoso Severini, L.; Alfonsi, R.; Mazzà, D.; Siler, M.; Coni, S.; Po, A.; et al. Itch/β-arrestin2-dependent non-proteolytic ubiquitylation of SuFu controls Hedgehog signalling and medulloblastoma tumorigenesis. Nat. Commun. 2018, 9, 976, doi:10.1038/s41467-018-03339-0.
- Tukachinsky, H.; Lopez, L.V.; Salic, A. A mechanism for vertebrate Hedgehog signaling: Recruitment to cilia and dissociation of SuFu-Gli protein complexes. J. Cell Biol. 2010, 191, 415–428, doi:10.1083/jcb.201004108.
- Yao, E.; Chuang, P.T. Hedgehog signaling: From basic research to clinical applications. J. Formos. Med. Assoc. 2015, 114, 569–576, doi:10.1016/j.jfma.2015.01.005.
- Montagnani, V.; Stecca, B. Role of Protein Kinases in Hedgehog Pathway Control and Implications for Cancer Therapy. Cancers 2019, 11, 449, doi:10.3390/cancers11040449.
- Liu, A. Proteostasis in the Hedgehog signaling pathway. Semin. Cell Dev. Biol. 2019, 93, 153–163, doi:10.1016/j.semcdb.2018.10.009.
- Hsia, E.Y.; Gui, Y.; Zheng, X. Regulation of Hedgehog signaling by ubiquitination. Front. Biol. (Beijing) 2015, 10, 203–220, doi:10.1007/s11515-015-1343-5.
- Niewiadomski, P.; Kong, J.H.; Ahrends, R.; Ma, Y.; Humke, E.W.; Khan, S.; Teruel, M.N.; Novitch, B.G.; Rohatgi, R. Gli protein activity is controlled by multisite phosphorylation in vertebrate Hedgehog signaling. Cell Rep. 2014, 6, 168–181, doi:10.1016/j.celrep.2013.12.003.
- Huntzicker, E.G.; Estay, I.S.; Zhen, H.; Lokteva, L.A.; Jackson, P.K.; Oro, A.E. Dual degradation signals control Gli protein stability and tumor formation. Genes Dev. 2006, 20, 276–281, doi:10.1101/gad.1380906.
- Di Marcotullio, L.; Ferretti, E.; Greco, A.; De Smaele, E.; Screpanti, I.; Gulino, A. Multiple ubiquitin-dependent processing pathways regulate hedgehog/gli signaling: Implications for cell development and tumorigenesis. Cell Cycle 2007, 6, 390–393, doi:10.4161/cc.6.4.3809.
- Gulino, A.; Di Marcotullio, L.; Canettieri, G.; De Smaele, E.; Screpanti, I. Hedgehog/Gli control by ubiquitination/acetylation interplay. Vitam. Horm. 2012, 88, 211–227, doi:10.1016/B978-0-12-394622-5.00009-2.
- Infante, P.; Lospinoso Severini, L.; Bernardi, F.; Bufalieri, F.; Di Marcotullio, L. Targeting Hedgehog Signalling through the Ubiquitylation Process: The Multiple Roles of the HECT-E3 Ligase Itch. Cells 2019, 8, 98, doi:10.3390/cells8020098.
- Di Marcotullio, L.; Ferretti, E.; Greco, A.; De Smaele, E.; Po, A.; Sico, M.A.; Alimandi, M.; Giannini, G.; Maroder, M.; Screpanti, I.; et al. Numb is a suppressor of Hedgehog signalling and targets Gli1 for Itch-dependent ubiquitination. Nat. Cell Biol. 2006, 8, 1415–1423, doi:10.1038/ncb1510.
- Di Marcotullio, L.; Greco, A.; Mazzà, D.; Canettieri, G.; Pietrosanti, L.; Infante, P.; Coni, S.; Moretti, M.; De Smaele, E.; Ferretti, E.; et al. Numb activates the E3 ligase Itch to control Gli1 function through a novel degradation signal. Oncogene 2011, 30, 65–76, doi:10.1038/onc.2010.394.
- Mazzà, D.; Infante, P.; Colicchia, V.; Greco, A.; Alfonsi, R.; Siler, M.; Antonucci, L.; Po, A.; De Smaele, E.; Ferretti, E.; et al. PCAF ubiquitin ligase activity inhibits Hedgehog/Gli1 signaling in p53-dependent response to genotoxic stress. Cell Death Differ. 2013, 20, 1688–1697, doi:10.1038/cdd.2013.120.
- Li, S.; Chen, Y.; Shi, Q.; Yue, T.; Wang, B.; Jiang, J. Hedgehog-regulated ubiquitination controls smoothened trafficking and cell surface expression in Drosophila. PLoS Biol. 2012, 10, e1001239, doi:10.1371/journal.pbio.1001239.
- Li, S.; Wang, B.; Jiang, J. Hedgehog reciprocally controls trafficking of Smo and Ptc through the Smurf family of E3 ubiquitin ligases. Sci. Signal. 2018, 11, doi:10.1126/scisignal.aan8660.
- Li, S.; Cho, Y.S.; Wang, B.; Jiang, J. Regulation of Smoothened ubiquitylation and cell surface expression through a Cul4-DDB1-Gβ E3 ubiquitin ligase complex. J. Cell Sci. 2018, 131, doi:10.1242/jcs.218016.
- Sun, X.; Sun, B.; Cui, M.; Zhou, Z. HERC4 exerts an anti-tumor role through destabilizing the oncoprotein Smo. Biochem. Biophys. Res. Commun. 2019, 513, 1013–1018, doi:10.1016/j.bbrc.2019.04.113.
- McMahon, A.P.; Ingham, P.W.; Tabin, C.J. Developmental roles and clinical significance of hedgehog signaling. Curr. Top. Dev. Biol. 2003, 53, 1–114, doi:10.1016/s0070-2153(03)53002-2.
- Ruiz i Altaba, A.; Sánchez, P.; Dahmane, N. Gli and hedgehog in cancer: Tumours, embryos and stem cells. Nat. Rev. Cancer 2002, 2, 361–372, doi:10.1038/nrc796.
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20, doi:10.17305/bjbms.2018.2756.
- Didiasova, M.; Schaefer, L.; Wygrecka, M. Targeting GLI Transcription Factors in Cancer. Molecules 2018, 23, 1003, doi:10.3390/molecules23051003.
- Peer, E.; Tesanovic, S.; Aberger, F. Next-Generation Hedgehog/GLI Pathway Inhibitors for Cancer Therapy. Cancers 2019, 11, 538, doi:10.3390/cancers11040538.
- Huang, S.Y.; Yang, J.Y. Targeting the Hedgehog Pathway in Pediatric Medulloblastoma. Cancers 2015, 7, 2110–2123, doi:10.3390/cancers7040880.
- Hutchin, M.E.; Kariapper, M.S.; Grachtchouk, M.; Wang, A.; Wei, L.; Cummings, D.; Liu, J.; Michael, L.E.; Glick, A.; Dlugosz, A.A. Sustained Hedgehog signaling is required for basal cell carcinoma proliferation and survival: Conditional skin tumorigenesis recapitulates the hair growth cycle. Genes Dev. 2005, 19, 214–223, doi:10.1101/gad.1258705.
- Scales, S.J.; de Sauvage, F.J. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol. Sci. 2009, 30, 303–312, doi:10.1016/j.tips.2009.03.007.
- Lee, Y.; Kawagoe, R.; Sasai, K.; Li, Y.; Russell, H.R.; Curran, T.; McKinnon, P.J. Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene 2007, 26, 6442–6447, doi:10.1038/sj.onc.1210467.
- De Smaele, E.; Di Marcotullio, L.; Ferretti, E.; Screpanti, I.; Alesse, E.; Gulino, A. Chromosome 17p deletion in human medulloblastoma: A missing checkpoint in the Hedgehog pathway. Cell Cycle 2004, 3, 1263–1266, doi:10.4161/cc.3.10.1200.
- Rubin, L.L.; de Sauvage, F.J. Targeting the Hedgehog pathway in cancer. Nat. Rev. Drug Discov. 2006, 5, 1026–1033, doi:10.1038/nrd2086.
- Mukherjee, S.; Frolova, N.; Sadlonova, A.; Novak, Z.; Steg, A.; Page, G.P.; Welch, D.R.; Lobo-Ruppert, S.M.; Ruppert, J.M.; Johnson, M.R.; et al. Hedgehog signaling and response to cyclopamine differ in epithelial and stromal cells in benign breast and breast cancer. Cancer Biol. Ther. 2006, 5, 674–683, doi:10.4161/cbt.5.6.2906.
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernández-del Castillo, C.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856, doi:10.1038/nature02009.
- Varnat, F.; Duquet, A.; Malerba, M.; Zbinden, M.; Mas, C.; Gervaz, P.; Ruiz i Altaba, A. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol. Med. 2009, 1, 338–351, doi:10.1002/emmm.200900039.
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat. Med. 2013, 19, 1410–1422, doi:10.1038/nm.3389.
- Fan, L.; Pepicelli, C.V.; Dibble, C.C.; Catbagan, W.; Zarycki, J.L.; Laciak, R.; Gipp, J.; Shaw, A.; Lamm, M.L.; Munoz, A.; et al. Hedgehog signaling promotes prostate xenograft tumor growth. Endocrinology 2004, 145, 3961–3970, doi:10.1210/en.2004-0079.
- Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N.P.; Guo, G.R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J.F.; et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat. Med. 2007, 13, 944–951, doi:10.1038/nm1614.
- Becher, O.J.; Hambardzumyan, D.; Fomchenko, E.I.; Momota, H.; Mainwaring, L.; Bleau, A.M.; Katz, A.M.; Edgar, M.; Kenney, A.M.; Cordon-Cardo, C.; et al. Gli activity correlates with tumor grade in platelet-derived growth factor-induced gliomas. Cancer Res. 2008, 68, 2241–2249, doi:10.1158/0008-5472.CAN-07-6350.
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172, doi:10.1016/j.cub.2006.11.033.
- Miele, E.; Po, A.; Begalli, F.; Antonucci, L.; Mastronuzzi, A.; Marras, C.E.; Carai, A.; Cucchi, D.; Abballe, L.; Besharat, Z.M.; et al. β-arrestin1-mediated acetylation of Gli1 regulates Hedgehog/Gli signaling and modulates self-renewal of SHH medulloblastoma cancer stem cells. BMC Cancer 2017, 17, 488, doi:10.1186/s12885-017-3477-0.
- Po, A.; Ferretti, E.; Miele, E.; De Smaele, E.; Paganelli, A.; Canettieri, G.; Coni, S.; Di Marcotullio, L.; Biffoni, M.; Massimi, L.; et al. Hedgehog controls neural stem cells through p53-independent regulation of Nanog. EMBO J. 2010, 29, 2646–2658, doi:10.1038/emboj.2010.131.
- Infante, P.; Alfonsi, R.; Botta, B.; Mori, M.; Di Marcotullio, L. Targeting GLI factors to inhibit the Hedgehog pathway. Trends Pharmacol. Sci. 2015, 36, 547–558, doi:10.1016/j.tips.2015.05.006.
- Quaglio, D.; Infante, P.; Di Marcotullio, L.; Botta, B.; Mori, M. Hedgehog signaling pathway inhibitors: An updated patent review (2015-present). Expert Opin. Ther. Pat. 2020, 30, 235–250, doi:10.1080/13543776.2020.1730327.
- Finley, D.; Bartel, B.; Varshavsky, A. The tails of ubiquitin precursors are ribosomal proteins whose fusion to ubiquitin facilitates ribosome biogenesis. Nature 1989, 338, 394–401, doi:10.1038/338394a0.
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479, doi:10.1146/annurev.biochem.67.1.425.
- Ye, Y.; Rape, M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764, doi:10.1038/nrm2780.
- Zhao, Y.; Sun, Y. Cullin-RING Ligases as attractive anti-cancer targets. Curr. Pharm. Des. 2013, 19, 3215–3225, doi:10.2174/13816128113199990300.
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012, 125, 531–537, doi:10.1242/jcs.091777.
- Walden, H.; Rittinger, K. RBR ligase-mediated ubiquitin transfer: A tale with many twists and turns. Nat. Struct. Mol. Biol. 2018, 25, 440–445, doi:10.1038/s41594-018-0063-3.
- Ciechanover, A.; Ben-Saadon, R. N-terminal ubiquitination: More protein substrates join in. Trends Cell Biol. 2004, 14, 103–106, doi:10.1016/j.tcb.2004.01.004.
- Pavri, R.; Zhu, B.; Li, G.; Trojer, P.; Mandal, S.; Shilatifard, A.; Reinberg, D. Histone H2B monoubiquitination functions cooperatively with FACT to regulate elongation by RNA polymerase II. Cell 2006, 125, 703–717, doi:10.1016/j.cell.2006.04.029.
- Su, Y.T.; Gao, C.; Liu, Y.; Guo, S.; Wang, A.; Wang, B.; Erdjument-Bromage, H.; Miyagi, M.; Tempst, P.; Kao, H.Y. Monoubiquitination of filamin B regulates vascular endothelial growth factor-mediated trafficking of histone deacetylase 7. Mol. Cell Biol. 2013, 33, 1546–1560, doi:10.1128/MCB.01146-12.
- Braten, O.; Livneh, I.; Ziv, T.; Admon, A.; Kehat, I.; Caspi, L.H.; Gonen, H.; Bercovich, B.; Godzik, A.; Jahandideh, S.; et al. Numerous proteins with unique characteristics are degraded by the 26S proteasome following monoubiquitination. Proc. Natl. Acad. Sci. USA 2016, 113, E4639–E4647, doi:10.1073/pnas.1608644113.
- Pickart, C.M. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533, doi:10.1146/annurev.biochem.70.1.503.
- Thrower, J.S.; Hoffman, L.; Rechsteiner, M.; Pickart, C.M. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000, 19, 94–102, doi:10.1093/emboj/19.1.94.
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229, doi:10.1146/annurev-biochem-060310-170328.
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422, doi:10.1038/cr.2016.39.
- Husnjak, K.; Dikic, I. Ubiquitin-binding proteins: Decoders of ubiquitin-mediated cellular functions. Annu. Rev. Biochem. 2012, 81, 291–322, doi:10.1146/annurev-biochem-051810-094654.
- Mevissen, T.E.; Hospenthal, M.K.; Geurink, P.P.; Elliott, P.R.; Akutsu, M.; Arnaudo, N.; Ekkebus, R.; Kulathu, Y.; Wauer, T.; El Oualid, F.; et al. OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell 2013, 154, 169–184, doi:10.1016/j.cell.2013.05.046.
- Nijman, S.M.; Luna-Vargas, M.P.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.; Sixma, T.K.; Bernards, R. A genomic and functional inventory of deubiquitinating enzymes. Cell 2005, 123, 773–786, doi:10.1016/j.cell.2005.11.007.
- Clague, M.J.; Urbé, S.; Komander, D. Breaking the chains: Deubiquitylating enzyme specificity begets function. Nat. Rev. Mol. Cell Biol. 2019, 20, 338–352, doi:10.1038/s41580-019-0099-1.
- Faesen, A.C.; Luna-Vargas, M.P.; Geurink, P.P.; Clerici, M.; Merkx, R.; van Dijk, W.J.; Hameed, D.S.; El Oualid, F.; Ovaa, H.; Sixma, T.K. The differential modulation of USP activity by internal regulatory domains, interactors and eight ubiquitin chain types. Chem. Biol. 2011, 18, 1550–1561, doi:10.1016/j.chembiol.2011.10.017.
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192, doi:10.1146/annurev-biochem-061516-044916.
- Urbé, S.; Liu, H.; Hayes, S.D.; Heride, C.; Rigden, D.J.; Clague, M.J. Systematic survey of deubiquitinase localization identifies USP21 as a regulator of centrosome- and microtubule-associated functions. Mol. Biol. Cell 2012, 23, 1095–1103, doi:10.1091/mbc.E11-08-0668.


