 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Darina Ocadlikova | + 3003 word(s) | 3003 | 2020-09-24 04:14:34 | | | |
| 2 | Rita Xu | -1357 word(s) | 1646 | 2020-09-28 11:55:04 | | |
Video Upload Options
The present investigation expands the knowledge on the immunogenic and tolerogenic potential of the chemotherapy drugs commonly used in the therapy of AML such as Daunorubicin, Cytarabine, Fludrabine and Etoposide . Among these, important differences have been observed, indicating that, particularly in an era when immunotherapy is being included in the clinical stage of AML treatment, the immunological perspective of chemotherapy should be taken into consideration in therapy decision-making.
1. Introduction
Recent evidence indicated that, under certain circumstances, chemotherapy stimulates the immune system. Indeed, in the last decade, some chemotherapeutic agents used for acute myeloid leukemia (AML) treatment, such as anthracyclines, have been shown to induce a type of cell death that can promote modifications in cancer cells, which activate the immune system against leukemia cells [1][2][3][4]. In particular, the treatment of AML cells with daunorubicin (DNR), but not cytarabine (Ara-C), results in the maturation of dendritic cells (DCs) and in the efficient cross-priming of anti-leukemia T cells [1][3][4][5][6][7]. This process, immunogenic cell death (ICD), is characterized by the coordinated emission of danger-associated molecular patterns (DAMPs), including the translocation of the endoplasmic reticulum (ER) chaperones such as calreticulin (CRT) and heat shock proteins (HSPs) 70 and 90 on the cell surface, the active secretion of adenosine triphosphate (ATP), the release of high mobility group box 1 (HMGB1) from the nucleus in the extracellular milieu [8][9][10][11][12] and, finally, the release of immunostimulatory cytokines, such as type I interferons [13][14]. In the early phase, CRT translocates from the ER to the outer leaflet of the plasma membrane, thus initiating the apoptotic caspase-dependent process [11][12]. Simultaneously, HSP70 and HSP90 bind tumor-associated antigens (TAAs), thus stimulating DC maturation [7][15]. During the late post-apoptotic phase, pro-inflammatory factor HMGB1—which binds the toll like receptor 4 (TLR4) on DCs—is released from the nucleus in the extracellular space [16]. Finally, autophagy-dependent active secretion of ATP—which binds purinergic receptors present on DCs—promotes the recruitment, survival and differentiation of DCs [17][18].
When emitted in the correct spatiotemporal context [12], these DAMPs recruit DCs in the proximity of ICD and activate them to engulf TAA processing and present them to CD4 and CD8 T cells, thus resulting in the priming of a robust, antigen-specific immune response [19][20].
Recent data support the role of chemotherapy in the activation of an anti-leukemia immune response [2][3][4][6][21], with important therapeutic implications. Indeed, Fredly et al. demonstrated that CRT is exposed by chemotherapy-treated primary human AML cells in 65% of tested patients. Moreover, in vitro cultured AML cells showed spontaneous release of HSP70 and 90 [6]. Recently, Fucikova et al. showed that CRT exposure by AML cells correlates with a strong anticancer immune response, improving the clinical outcomes of AML patients [3][22]. Prognostic statistical analysis of CRT, HSP70 and HSP 90 exposure revealed that ICD-associated DAMPs correlate with improved disease outcomes in patients with AML [3]. Wemeau et al. also demonstrated that CRT exposure on malignant blasts predicts a cellular anticancer immune response in patients with AML [4].
Although the scenario of AML is rapidly evolving and new targeted drugs are entering the clinical stage [23][24][25], the therapy of AML is at present principally based on the use of cytotoxic and cytostatic drugs, such as anthracyclines [26][27][28] and Ara-C [27][29][30]. Nevertheless, in induction chemotherapy, the conventional treatment regimen is able to induce complete remission (CR) in up to 70% of adult patients [31][32]. However, the probability of relapse remains elevated, particularly in elderly or prognostically “high risk” patients, unless transplantation of autologous or allogeneic hematopoietic stem cells is performed as a post-CR consolidation strategy [33].
2. DNR, Eto, Ara-C and Flu Induced a Similar Apoptosis Level in HL-60, KG-1 and Primary AML Cells
Before evaluating the immunogenic effect of tested chemotherapy drugs such as DNR, Eto, Ara-C and Flu, their cytotoxic activity on HL-60, KG-1 and primary AML cells was assessed to select the concentration of each drug capable of inducing similar and comparable levels of apoptosis. Various concentrations of each drug were tested. In particular, 10 µg/mL, 20 µg/mL, 50 µg/mL and 100 µg/mL for Eto, and 10 µg/mL, 20 µg/mL, 50 µg/mL, 70 µg/mL and 100 µg/mL for Flu were used (data not shown) to achieve an apoptosis level comparable to that previously determined for DNR and Ara-C [1]. As shown in Figure 1, the concentrations which induced a similar apoptosis level (percentage of annexin-V+ cells) to DNR and Ara-C treatment, were the following: 20 µg/mL for Eto with 53.9 ± 8.6%/62.4 ± 5.5%/43.2 ± 6.3% (HL-60/KG-1/primary AML cells, respectively), and 70 µg/mL for Flu with 24 ± 2.9%/31 ± 7.3%/39.4 ± 2.4%, of Annexin-V+ cells, respectively (mean ± SEM), as compared to 43.2 ± 3.1%/49.4 ± 5.7%/49.8 ± 4.3% for DNR at the concentration of 500 ng/mL, and 46.5 ± 39%/52 ± 7.1%/48.6 ± 3.6% for Ara-C at the concentration of 20 µg/mL.
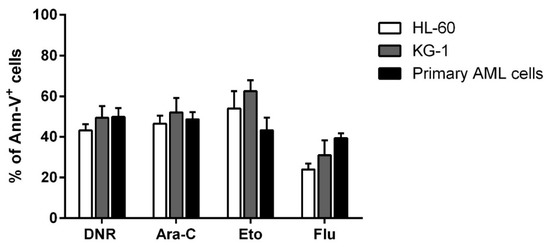
Figure 1. Flow cytometry analysis of acute myeloid leukemia (AML) cell apoptosis after chemotherapy treatment. The HL-60, KG-1 and primary AML cells were treated with daunorubicin (DNR) (500 ng/mL), cytarabine (Ara-C) (20 µg/mL), etoposide (Eto) (20 µg/mL) or fludarabine (Flu) (70 µg/mL) for 24 h. The percentage of apoptotic Ann-V+ cells was assessed by flow cytometry. The values are calculated as differences between treated and un-treated cells and represented as mean ± SEM of 5 independent experiments.
3. Only Treatment with DNR and Eto, but Not Flu and Ara-C, Induced Translocation of CRT and HSPs from ER to Plasma Cell Membrane in AML Cells
ICD is represented by the coordinated emission of DAMPs, including CRT and HSPs 70 and 90 translocation from the ER to the cell surface, the active secretion of ATP and the active release of HMGB1 from the nucleus to the extracellular milieu [12]. We then in vitro tested and compared the capacity of each drug to induce these events. First, the evaluation of CRT, HSP70 and HSP90 exposure was determined in apoptotic cells by flow cytometry. HL-60, KG-1 or primary AML cells were treated with DNR (500 ng/mL), Ara-C (20 µg/mL), Eto (20 µg/mL) or Flu (70 µg/mL) for 24 h. The protein expression levels obtained by flow cytometry varied considerably among the different conditions (Figure 2). In particular, in the case of CRT, the expression significantly increased from 0.6 ± 0.1%/2.4 ± 0.4%/4 ± 1.8% (HL-60/KG-1/primary AML cells, respectively) in un-treated cells to 39.7 ± 9.7%/37.6 ± 4.9%/22.8 ± 5.5% in DNR-treated cells (p < 0.0001/0.001/0.001, respectively), or 35.5 ± 9.7%/33.5 ± 0.3%/22.2 ± 3.5% in Eto-treated cells (p < 0.001/0.01/0.05, respectively). In contrast, no significant differences were observed when Ara-C (2.9 ± 0.3%/4.3 ± 1.9%/3.3 ± 0.7%) or Flu (3.7 ± 1.5%/3 ± 2%/5.8 ± 3.5%) were used. Similarly, a significant HSP exposure was observed for HL-60, KG-1 and primary AML cells after DNR and Eto treatment, but not after Ara-C and Flu treatment (Figure 2). The only exception to this trend was represented by primary AML cells for HSP90 protein, where no up-regulation after Eto treatment was observed.
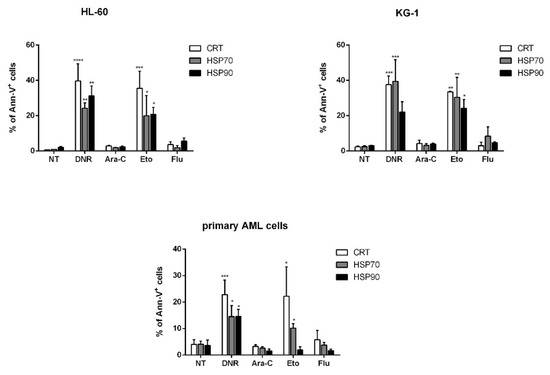
Figure 2. Flow cytometry analysis of calreticulin (CRT) and heat-shock protein (HSP) translocation on the cell-surface of acute myeloid leukemia (AML) cells after chemotherapy treatment. The HL-60, KG-1 and primary AML cells were treated with daunorubicin (DNR) (500 ng/mL), cytarabine (Ara-C) (20 µg/mL), etoposide (Eto) (20 µg/mL) and fludarabine (Flu) (70 µg/mL) for 24 h. The percentage of CRT+, HSP70+ and HSP90+ cells (gated on apoptotic Ann-V+ cells) was analyzed by flow cytometry. Un-treated cells (no treatment; NT) were used as a negative control. The values are represented as mean ± SEM of 5 independent experiments. * p <0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001 compared to un-treated cells.
As shown in Figure 3, flow cytometry data regarding CRT translocation were also confirmed using immunofluorescence. Taken together, these in vitro data indicate that Eto is comparable to DNR in early ICD event induction. In contrast, Flu treatment is not capable of inducing either CRT or HSP translocation, similarly to Ara-C treatment.
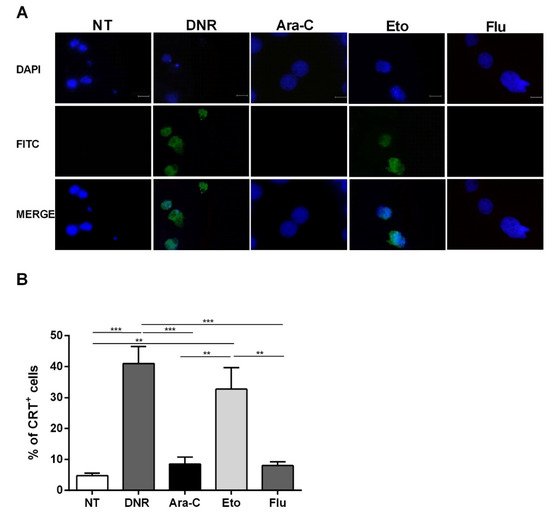
Figure 3. Immunofluorescence analysis of calreticulin (CRT) translocation on the cell-surface of HL-60 cell lines after chemotherapy treatment. The HL-60 cells were treated or not (no treatment; NT) with daunorubicin (DNR) (500 ng/mL), cytarabine (Ara-C) (20 µg/mL), etoposide (Eto) (20 µg/mL) and fludarabine (Flu) (70 µg/mL) for 24 h. (A) The localization of CRT (FITC-conjugated) at cell membrane level with respect to the nucleus (DAPI-conjugated) was evaluated by immunofluorescence microscopy. One representative experiment for each drug is reported. Bar 20 µm. (B) Quantitative analysis of CRT+ cells by immunofluorescence. A total of 100 cells were used for the quantification. ** p < 0.01; *** p < 0.001.
4. Only Treatment with DNR and Eto, but Not Flu and Ara-C, Induced the HMGB1 Release from the Nucleus to the Extracellular Space of AML Cells
After cellular stress, during the late post-apoptotic phase, pro-inflammatory nuclear factor HMGB1 translocates to the cytosol and is consequently released to the extracellular space [12]. After binding to specific receptors on DCs, HMGB1 induces the full maturation of DCs as evaluated by the up-regulation of CD40, CD54, CD80, CD83 and MHC II. To test this event, the HL-60 cells were treated with DNR (500 ng/mL), Ara-C (20 µg/mL), Eto (20 µg/mL) or Flu (70 µg/mL) for 24 h, then fixed, permeabilized, stained and analyzed for HMGB1 expression by immunofluorescence microscopy. As shown in Figure 4A, the extracellular release of HMGB1 was well-documented after DNR and Eto, but not after Ara-C and Flu, similarly to what was observed for CRT and HSP induction. These results were also confirmed when the HMGB1 expression density between un-treated and treated cells was evaluated (Figure 4B).
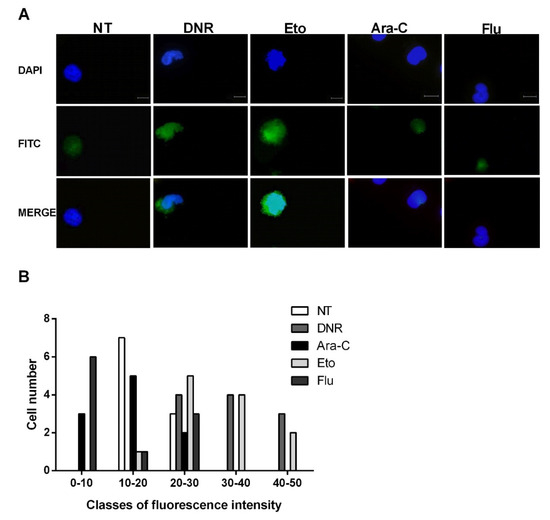
Figure 4. Immunofluorescence analysis of non-histone chromatin-binding protein high mobility group box 1 (HMGB1) release from the nucleus of HL-60 cell lines after chemotherapy treatment. The HL-60 cells were treated or not (no treatment; NT) with daunorubicin (DNR) (500 ng/mL), cytarabine (Ara-C) (20 µg/mL), etoposide (Eto) (20 µg/mL) and fludarabine (Flu) (70 µg/mL) for 24 h. (A) The release of HMGB1 (FITC-conjugated) from nucleus (DAPI-conjugated) to cytoplasm and then extracellular space was visualized by immunofluorescence microscopy. One representative experiment for each drug is reported. Bar 20 µm. (B) Quantitative analysis of HMGB1 fluorescence intensity outside the nucleus in un-treated and treated HL-60 cells. A representative field was used for quantification. The signal outside the nucleus was measured by densitometry (n = 21; randomly selected cells). The cells are grouped in classes of fluorescence intensity and plotted relative to HMGB1 expression.
Collectively, in line with early ICD-related events, the immunofluorescence evaluation confirmed the presence of HMGB1 in the extracellular milieu after DNR (as expected) and Eto treatment, but not after Flu and Ara-C.
References
- Lecciso, M.; Ocadlikova, D.; Sangaletti, S.; Trabanelli, S.; De Marchi, E.; Orioli, E.; Pegoraro, A.; Portararo, P.; Jandus, C.; Bontadini, A.; et al. ATP Release from Chemotherapy-Treated Dying Leukemia Cells Elicits an Immune Suppressive Effect by Increasing Regulatory T Cells and Tolerogenic Dendritic Cells. Front. Immunol. 2017, 8, 1918, doi:10.3389/fimmu.2017.01918.
- Fucikova, J.; Kralikova, P.; Fialova, A.; Brtnicky, T.; Rob, L.; Bartunkova, J.; Spísek, R. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res. 2011, 71, 4821–4833, doi:10.1158/0008-5472.CAN-11-0950.
- Fucikova, J.; Truxova, I.; Hensler, M.; Becht, E.; Kasikova, L.; Moserova, I.; Vosahlikova, S.; Klouckova, J.; Church, S.E.; Cremer, I.; et al. Calreticulin exposure by malignant blasts correlates with robust anticancer immunity and improved clinical outcome in AML patients. Blood 2016, 128, 3113–3124, doi:10.1182/blood-2016-08-731737.
- Wemeau, M.; Kepp, O.; Tesnière, A.; Panaretakis, T.; Flament, C.; De Botton, S.; Zitvogel, L.; Kroemer, G.; Chaput, N. Calreticulin exposure on malignant blasts predicts a cellular anticancer immune response in patients with acute myeloid leukemia. Cell Death Dis. 2010, 1, e104, doi:10.1038/cddis.2010.82.
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72, doi:10.1146/annurev-immunol-032712-100008.
- Fredly, H.; Ersvær, E.; Gjertsen, B.T.; Bruserud, O. Immunogenic apoptosis in human acute myeloid leukemia (AML): Primary human AML cells expose calreticulin and release heat shock protein (HSP) 70 and HSP90 during apoptosis. Oncol. Rep. 2011, 25, 1549–1556, doi:10.3892/or.2011.1229.
- Spisek, R.; Charalambous, A.; Mazumder, A.; Vesole, D.H.; Jagannath, S.; Dhodapkar, M.V. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: Therapeutic implications. Blood 2007, 109, 4839–4845, doi:10.1182/blood-2006-10-054221.
- Garg, A.D.; Martin, S.; Golab, J.; Agostinis, P. Danger signalling during cancer cell death: Origins, plasticity and regulation. Cell Death Differ. 2014, 21, 26–38, doi:10.1038/cdd.2013.48.
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059, doi:10.1038/nm1622.
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577, doi:10.1126/science.1208347.
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61, doi:10.1038/nm1523.
- Garg, A.D.; Dudek, A.M.; Agostinis, P. Cancer immunogenicity, danger signals, and DAMPs: What, when, and how? Biofactors 2013, 39, 355–367, doi:10.1002/biof.1125.
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178, doi:10.1038/nm.2028.
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remédios, C.; et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309, doi:10.1038/nm.3708.
- Obeid, M.; Tesniere, A.; Panaretakis, T.; Tufi, R.; Joza, N.; van Endert, P.; Ghiringhelli, F.; Apetoh, L.; Chaput, N.; Flament, C.; et al. Ecto-calreticulin in immunogenic chemotherapy. Immunol. Rev. 2007, 220, 22–34, doi:10.1111/j.1600-065X.2007.00567.x.
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Criollo, A.; Ortiz, C.; Lidereau, R.; Mariette, C.; Chaput, N.; Mira, J.P.; Delaloge, S.; et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol. Rev. 2007, 220, 47–59, doi:10.1111/j.1600-065X.2007.00573.x.
- Ma, Y.; Adjemian, S.; Yang, H.; Catani, J.P.; Hannani, D.; Martins, I.; Michaud, M.; Kepp, O.; Sukkurwala, A.Q.; Vacchelli, E.; et al. ATP-dependent recruitment, survival and differentiation of dendritic cell precursors in the tumor bed after anticancer chemotherapy. Oncoimmunology 2013, 2, e24568, doi:10.4161/onci.24568.
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282–286, doi:10.1038/nature08296.
- Cirone, M.; Di Renzo, L.; Lotti, L.V.; Conte, V.; Trivedi, P.; Santarelli, R.; Gonnella, R.; Frati, L.; Faggioni, A. Activation of dendritic cells by tumor cell death. Oncoimmunology 2012, 1, 1218–1219, doi:10.4161/onci.20428.
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875, doi:10.1038/nrc3380.
- Chen, X.; Fosco, D.; Kline, D.E.; Kline, J. Calreticulin promotes immunity and type I interferon-dependent survival in mice with acute myeloid leukemia. Oncoimmunology 2017, 6, e1278332, doi:10.1080/2162402X.2016.1278332.
- Fucikova, J.; Kasikova, L.; Truxova, I.; Laco, J.; Skapa, P.; Ryska, A.; Spisek, R. Relevance of the chaperone-like protein calreticulin for the biological behavior and clinical outcome of cancer. Immunol. Lett. 2018, 193, 25–34, doi:10.1016/j.imlet.2017.11.006.
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117, doi:10.1158/2159-8290.CD-16-0313.
- Alfayez, M.; Borthakur, G. Checkpoint inhibitors and acute myelogenous leukemia: Promises and challenges. Expert Rev. Hematol. 2018, 11, 373–389, doi:10.1080/17474086.2018.1459184.
- Hobo, W.; Hutten, T.J.A.; Schaap, N.P.M.; Dolstra, H. Immune checkpoint molecules in acute myeloid leukaemia: Managing the double-edged sword. Br. J. Haematol. 2018, 181, 38–53, doi:10.1111/bjh.15078.
- Bradstock, K.F.; Link, E.; Di Iulio, J.; Szer, J.; Marlton, P.; Wei, A.H.; Enno, A.; Schwarer, A.; Lewis, I.D.; D’Rozario, J.; et al. Idarubicin Dose Escalation During Consolidation Therapy for Adult Acute Myeloid Leukemia. J. Clin. Oncol. 2017, 35, 1678–1685, doi:10.1200/JCO.2016.70.6374.
- Burnett, A.K.; Russell, N.H.; Hills, R.K.; Kell, J.; Cavenagh, J.; Kjeldsen, L.; McMullin, M.F.; Cahalin, P.; Dennis, M.; Friis, L.; et al. A randomized comparison of daunorubicin 90 mg/m2 vs 60 mg/m2 in AML induction: Results from the UK NCRI AML17 trial in 1206 patients. Blood 2015, 125, 3878–3885, doi:10.1182/blood-2015-01-623447.
- Balakrishnan, V.S. Escalated anthracycline dose in adult AML. Lancet Oncol. 2017, 18, e253, doi:10.1016/S1470-2045(17)30277-2.
- Magina, K.N.; Pregartner, G.; Zebisch, A.; Wölfler, A.; Neumeister, P.; Greinix, H.T.; Berghold, A.; Sill, H. Cytarabine dose in the consolidation treatment of AML: A systematic review and meta-analysis. Blood 2017, 130, 946–948, doi:10.1182/blood-2017-04-777722.
- Murphy, T.; Yee, K.W.L. Cytarabine and daunorubicin for the treatment of acute myeloid leukemia. Expert Opin. Pharmacother. 2017, 18, 1765–1780, doi:10.1080/14656566.2017.1391216.
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447, doi:10.1182/blood-2016-08-733196.
- Estey, E. Acute myeloid leukemia: 2016 Update on risk-stratification and management. Am. J. Hematol. 2016, 91, 824–846, doi:10.1002/ajh.24439.
- Burnett, A.; Wetzler, M.; Löwenberg, B. Therapeutic advances in acute myeloid leukemia. J. Clin. Oncol. 2011, 29, 487–494, doi:10.1200/JCO.2010.30.1820.


