 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Carlos Alonso-Moreno | + 1938 word(s) | 1938 | 2020-09-02 15:27:41 | | | |
| 2 | Felix Wu | Meta information modification | 1938 | 2020-10-29 03:18:03 | | |
Video Upload Options
Nanoparticles (NPs) are promising drug delivery systems (DDS) for identifying and treating cancer. Active targeting NPs can be generated by conjugation with ligands that bind overexpressed or mutant cell surface receptors on target cells that are poorly or not even expressed on normal cells. Receptor-mediated endocytosis of the NPs occurs and the drug is released inside the cell or in the surrounding tissue due to the bystander effect. Antibodies are the most frequently used ligands to actively target tumor cells. In this context, antibody-based therapies have been extensively used in HER2+ breast cancer.
1. Introduction
The antibody conjugated NPs (ACNPs) approach is built on the success of nanotechnology and antibody therapies. When compared with the development of antibody drug conjugates (ADCs), ACNPs present many advantages, such as the delivery of the drug in a controlled manner, preservation of the chemical structure of the drug, reduced risk of secondary metabolites (if metabolism is unpredictable), and finally, potentially reduced toxicity [1]. Based on these features, the membrane proteins overexpressed in tumor cells can be used to design the antibodies that will later be implemented as the nanoparticle vector. The mechanism of action of both targeted therapies (ADCs and ACNPs) are very similar. After binding to the target, the complexes are internalized into the cell through receptor-mediated endocytosis [2], resulting in the formation of endosomes. Later, endosomes and lysosomes are coupled to release the drug into the cytoplasm [3]. However, the cargo of the nanoparticle can also diffuse directly through the cellular membrane, augmenting the cellular penetration of the compound.
2. Polymeric Antibody Conjugated Nanoparticles (ACNPs) to Improve Treatments in Breast Cancer
Polymeric NPs appear to be the most promising drug carriers due to their nanoscale size and potential for selective targeting and controlled drug release [4][5][6][7]. NPs in the range of 100–400 nm have been widely reported to accumulate at the tumor site through the enhanced permeability and retention (EPR) effect [8][9]. This favors high accumulation of the drug, facilitating its delivery to the site of interest by convection and diffusion processes [10], reducing, at the same time, the damage to surrounding tissues [11][12]. The passive targeting is drastically influenced by the size and surface charge of the NPs. Both features are important for the retention of the NP in the tumor and for its circulation time.
After accumulation in the tumor region, drug efficiency can even be increased by the so-called active targeting. Binding to overexpressed receptors on target cells [13] that are poorly or not even expressed on normal cells [14][15][16][17] affords active targeting of NPs to tumor cells. Actively targeted NPs allow the delivery of the drugs at the desired location, avoiding normal tissues and, therefore, enhancing the therapeutic efficiency. For example, NPs bearing the monoclonal antibody trastuzumab, which interacts with the surface protein HER2, are expected to accumulate at sites where HER2 is overexpressed, such as in breast or gastric tumors. These particles can be loaded with other agents such as taxanes, which synergize with trastuzumab in the treatment of breast tumors. NP internalization occurs via receptor-mediated endocytosis [17][18]. Antibodies are the most frequently used ligands to target tumor cells [19][20]. Antibody fragments, such as antigen-binding fragments (Fab) may also be conjugated to reach higher diffusion rates and improve tumor uptake [21][22]. Trastuzumab is the most widely explored monoclonal antibody for generating novel nanomedicines for breast cancer therapy by far.
3. Conjugation Strategies for ACNP Generation
NP functionalization with antibodies or antibody fragments can be carried out mainly via the adsorption phenomenon, covalent-nature binding, or binding by the use of adapter molecules (Figure 1). The immobilization of antibodies into the nanoparticle surface must guarantee both the desired amount of antibodies per nanoparticle and the proper antibody orientation [23][24]. Additionally, the immobilization method must generate a stable bond and preserve the biological activity of the antibody [25].
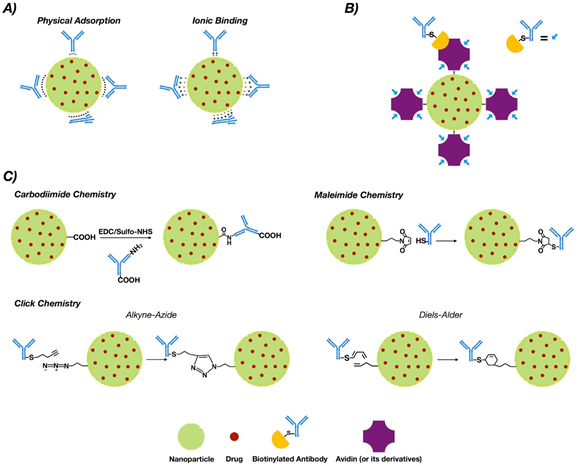
Figure 1. Conjugation strategies for antibody conjugated nanoparticle (ACNP) generation: (A) adsorption; (B) use of adapters; and (C) covalent binding.
3.1. Adsorption
This phenomenon is a non-covalent immobilization strategy that comprises physical adsorption and ionic binding [26] (see Figure 1A). Physical adsorption occurs via weak interactions such as hydrogen bonding, electrostatic, hydrophobic and Van der Waals attractive forces [27], while ionic binding occurs between the opposite charges of the antibodies and NP surfaces [28]. However, when compared to other methodologies such as covalent binding, adsorption provides less stability [29]. On the other hand, the fact that the interaction is non-covalent may allow release of the antibody in the tumor tissue, allowing deploy of its antitumoral properties. In this regard, three modified methodologies of functionalized PLGA NPs with trastuzumab were generated via adsorption, charged interactions and covalent binding. The results obtained via covalent binding demonstrated its higher stability versus charge interactions and adsorption [30].
3.2. Covalent Strategies
Covalent binding requires prior activation of the NPs [31]. Commonly, covalent strategies occur via carbodiimide chemistry, maleimide chemistry or “click chemistry”.
3.2.1. Chemistry of Carbodiimide
The most common covalent conjugation corresponds with the binding through the primary amines of the antibodies [32] (see Figure 1C), without performing any chemical modification [33]. Previously, carboxyl groups of the NPs must be activated by the addition of cross-linking agents, where 1-ethyl-3-(-3-dimethylaminopropyl) carbodiimide (EDC) is the most used [34][35]. Even though it is not required, N-hydroxysuccinimide (NHS) or N-hydroxysulfosuccinimide (sulfo-NHS) are usually added to improve the coupling efficiency [36][37].
This methodology is easy but the coupling between functional groups and crosslinkers is not selective. This absence of control over antibody orientation into the NP surface is a primary disadvantage [38][39].
3.2.2. Maleimide Chemistry
This approach occurs through sulfhydryl groups (–SH) of antibodies (Figure 1C). Sulfhydryl groups are much less abundant than primary amines or thiols. They are present in the side chain of cysteine [40]. Free sulfhydryl, the de-protonated form of thiol at physiological pH, is a better nucleophile than thiol groups [41]. The –SH groups are generated on the antibodies via reaction with primary amines or by reduction of native disulfide bonds of antibodies [42][43].Furthermore, this chemical group can be easily obtained by modification of the ε-amino of lysine residues with sulfhydryl-addition reagents. 2-Iminothiolane (Traut’s reagent) and N-succinimidyl S-acetylthioacetate (SATA) are the most common [44].
The reaction of maleimide groups towards free sulfhydryls is a thousand times faster than with primary amines at neutral pH, which improves selectivity [45][46]. A stable thioether linkage is formed after alkylation reaction with sulfhydryl groups [47][48] (see Figure 1C). The most used maleimide cross-linking reagents are the NHS/maleimide heterobifunctional linkers, PEGylated analogues (NHS-PEG-maleimide), succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC) and sulfosuccinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (sulfo-SMCC) [49][50][51]. NHS-PEG-maleimide has been the most extensively used crosslinker in antibody conjugation strategies in polymeric NPs for breast cancer treatment. SMCC is water-insoluble [52] but sulfo-SMCC is soluble in water due to a negatively charged sulfonate group on the NHS ring [53]. Therefore, using sulfo-SMCC as crosslinker, the free sulfhydryls of the antibody can be linked to the primary amines on the surface of the NPs via maleimide chemistry [54]. In this way, trastuzumab was attached to self-assembled chitosan-doxorubicin NPs via thiolation of lysine residues and subsequent linking of the thiols through sulfo-SMCC crosslinker to chitosan [55]. These ACNPS discriminated between HER2+ and HER2− cells in their mechanism of action. Heterobifunctional PEGylated linkers can also be used in the conjugation of thiolated antibodies to NPs through maleimide chemistry [56][57].
3.2.3. Click Chemistry
“Click chemistry” reactions occur efficiently at room temperature, under mild and in aqueous solvents. These reactions, which require no or minimal purification, resulted in irreversible chemical bonds with the absence of cytotoxic byproducts [58][59][60][61].
[3+2] Azide–alkyne cycloaddition (AAC) reactions catalyzed by copper (I) (CuAAC), strain-promoted [3+2] azide–alkyne cycloaddition (SPAAC) reactions, typical [4+2] Diels–Alder (DA), and inverse electron demand hetero Diels–Alder (iEDDA) reactions [62][63][64][65][66] configure the “click chemistry” strategy. The advantage of using azide and alkyne groups relys on their low existence in biological systems and inertness towards the majority of functional groups and biomolecules [67]. Functionalization of NPs with azide or alkyne moieties goes through EDC/NHS coupling or maleimide–thiol conjugation. In this context, NHS ester or maleimide must be present at one side of the linker, while azide or alkyne groups must be located at the other [68].
Cu(I) catalysts accelerate AAC reactions [69][70]. However, the toxicity associated with these catalysts holds back their use in living systems [71]. Thus, CuAAC reactions were used to develop a HER2 targeted ACNP for delivering DOX to breast cancer cells [72]. These ACNPs included a pH sensitive block polymer, poly(L-histidine). A NHS-PEG-alkyne linker was introduced to trastuzumab to provide an alkyne group into the antibody and thus further improve cellular uptake and toxicity for MCF7 and SKBR3 breast cancer cells. In the same way, optimum polyethylenglycol (PEG) coverage over trastuzumab conjugated ACNPs was also prepared via CuAAC conjugation to improve the pharmacokinetics [73]. Of note, developments in copper-free variants of AAC were achieved by introducing a strain-promoted azide–alkyne cycloaddition (SPAAC) reaction to overcome the CuAAC limitations [74][75][76].
The SPAAC variant introduces ring-strained alkynes (cycloalkynes) to create stable triazoles [77]. Conjugated PLGA–PEG NPs with Fab fragments of trastuzumab or cetuximab for the treatment of breast cancer were obtained via SPAAC [78]. The evaluation of these ACNPs using HER2+ breast cancer lines (HCC1954) demonstrated that site-specific conjugation by SPAAC increased the antigen binding capacity and the conjugation efficiency [78].
3.3. Binding by Adapter Molecules
Non-covalent approaches by using adapter biomolecules avoid randomly oriented antibodies [79]. The most relevant binding strategy exploits the biotin–avidin interaction, which is based on the strong binding affinity between biotin and a biotin-binding protein, such as avidin or its analogues [80][81] (See Figure 1B).
Biotin is a potential tumor-targeting moiety because it is a water-soluble vitamin (vitamin H), it is essential for growth and cell signaling, and their receptors are overexpressed on a broad range of cancer cells [82][83][84]. As an example of this non-covalent approach, the biotin-decorated PEG-PCL NPs were co-loaded with doxorubicin and quercetin to overcome doxorubicin-resistant breast cancer. In the same way, 7-ethyl-10-hydroxycamptothecin (SN-38) was successfully loaded into PLGA-PEG-biotin NPs to treat breast cancer. Compared to non-biotin conjugated NPs, these conjugated NPs increased the in vivo antitumor drug efficacy [85][86].
On the other hand, three N-acetyl glucosamine and four mannose residues constitute the oligosaccharide side chain of the tetrameric glycoprotein avidin [87][88]. Each of the four avidin monomers can bind to biotin [89]. However, because avidin is a basic protein with a high isoelectric point (pI) (~10.5) [90], both positively charged residues at physiological pH and glycosylation may lead to nonspecific binding of avidin to other molecules [91]. Thus, other forms of avidin, like streptavidin or neutravidin, are preferred [92]. Contrary to avidin, streptavidin is not a glycoprotein [93][94] and its pI is much lower, which prevents interactions with sugar receptors [95]. One of the most elegant strategies applied consisted in engineering monomeric avidin/streptavidin to monovalent biotin binding [96] and nanosized poly-avidins named Avidin-Nucleic-Acid-Nano-Assemblies (ANANAS) [97]. Notably, the ANANAS technology along with the use of an antibody (cetuximab) to target a ligand demonstrated an in vivo therapeutic efficacy at a dose of doxorubicin below the one clinically used [98].
To minimize the immunogenicity, a recombinant non-glycosylated form of avidin (neutravidin), was developed [99][100]. Warlick et al. prepared anti-HER2-modified NPs by the use of biotin/neutravidin aptamers to efficiently bind to and internalize in HER2-overexpressing cells. Trastuzumab was biotinylated and human serum albumin NPs were functionalized with neutravidin via NHS-PEG-Mal heterobifunctional crosslinker conjugation. This work clearly proved ACNPs combine specific tumor targeting with the drug delivery properties[101] .
Finally, the use of aptamers was extended by Powell et al. into the development of a targeted delivery of siRNA specific to the multidrug resistance (MDR) gene of metastatic breast cancer. The use of a cholesterol, DOTAP and PLGA or PLGA-PEG lipid-polymer hybrid liposomes nanocarrier system overcame resistances of breast cancer cells generated against therapeutic drugs. The mechanism consists in enhancing the knockdown of the MDR gene (P-gp) by the use of aptamer-labeled P-gp siRNA encapsulated NPs [102].
References
- Johnston, M.C.; Scott, C.J. Antibody Conjugated Nanoparticles as a Novel Form of Antibody Drug Conjugate Chemotherapy. Drug Discov. Today: Technol. 2018, 30, 63–69.
- Chari, R.V.J. Targeted cancer therapy: Conferring specificity to cytotoxic drugs. Acc. Chem. Res. 2008, 41, 98–107.
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. MAbs 2014, 6, 34–45.
- Duncan, R.; Gaspar, R. Nanomedicine(s) under the Microscope. Mol. Pharm. 2011, 8, 2101–2141.
- Malaspina, D.C.; Longo, G.; Szleifer, I. Behavior of Ligand Binding Assays with Crowded Surfaces: Molecular Model of Antigen Capture by Antibody-Conjugated Nanoparticles. PLoS ONE 2017, 12, e0185518.
- Steichen, S.D.; Caldorera-Moore, M.; Peppas, N.A. A Review of Current Nanoparticle and Targeting Moieties for the Delivery of Cancer Therapeutics. Eur. J. Pharm. Sci. 2013, 48, 416–427.
- Danhier, F.; Feron, O.; Préat, V. To Exploit the Tumor Microenvironment: Passive and Active Tumor Targeting of Nanocarriers for Anti-Cancer Drug Delivery. J. Control. Release 2010, 148, 135–146.
- Bawa, R. FDA and Nanotech: Baby Steps Lead to Regulatory Uncertainty. In Bio-Nanotechnology; Blackwell Publishing Ltd.: Oxford, UK, 2013; pp. 720–732.
- Nakamura, Y.; Mochida, A.; Choyke, P.L.; Kobayashi, H. Nanodrug Delivery: Is the Enhanced Permeability and Retention Effect Sufficient for Curing Cancer? Bioconjugate Chem. 2016, 27, 2225–2238.
- Andreas Wicki; Dominik Witzigmann; Vimalkumar Balasubramanian; Jörg Huwyler; Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. Journal of Controlled Release 2015, 200, 138-157, 10.1016/j.jconrel.2014.12.030.
- Nichols, J.W.; Bae, Y.H. Odyssey of a Cancer Nanoparticle: From Injection Site to Site of Action. Nano Today 2012, 7, 606–618.
- Xin, Y.; Yin, M.; Zhao, L.; Meng, F.; Luo, L. Recent Progress on Nanoparticle-Based Drug Delivery Systems for Cancer Therapy. Cancer Biol. Med. 2017, 14, 228–241.
- Changyou Zhan; Juan Guan; Jun Qian; Changyou Zhan; Peptide ligand-mediated targeted drug delivery of nanomedicines. Biomaterials Science 2019, 7, 461-471, 10.1039/c8bm01340c.
- Allen, T.M. Ligand-Targeted Therapeutics in Anticancer Therapy. Nat. Rev. Cancer 2002, 2, 750–763.
- Stefanick, J.F.; Omstead, D.T.; Kiziltepe, T.; Bilgicer, B. Dual-Receptor Targeted Strategy in Nanoparticle Design Achieves Tumor Cell Selectivity through Cooperativity. Nanoscale 2019, 11, 4414–4427.
- Ud Din, F.; Aman, W.; Ullah, I.; Qureshi, O.S.; Mustapha, O.; Shafique, S.; Zeb, A. Effective Use of Nanocarriers as Drug Delivery Systems for the Treatment of Selected Tumors. Int. J. Nanomed. 2017, 12, 7291–7309.
- Pirollo, K.F.; Chang, E.H. Does a Targeting Ligand Influence Nanoparticle Tumor Localization or Uptake? Trends Biotechnol. 2008, 26, 552–558. [
- Lisa M. Bareford; Peter W. Swaan; Endocytic mechanisms for targeted drug delivery☆. Advanced Drug Delivery Reviews 2007, 59, 748-758, 10.1016/j.addr.2007.06.008.
- Bregoli, L.; Movia, D.; Gavigan-Imedio, J.D.; Lysaght, J.; Reynolds, J.; Prina-Mello, A. Nanomedicine Applied to Translational Oncology: A Future Perspective on Cancer Treatment. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 81–103.
- Farahavar, G.; Abolmaali, S.S.; Gholijani, N.; Nejatollahi, F. Antibody-Guided Nanomedicines as Novel Breakthrough Therapeutic, Diagnostic and Theranostic Tools. Biomater. Sci. 2019, 7, 4000–4016.
- Zhang, B.; Hu, Y.; Pang, Z. Modulating the Tumor Microenvironment to Enhance Tumor Nanomedicine Delivery. Front. Pharmacol. 2017, 8, 952.
- Xenaki, K.T.; Oliveira, S.; van Bergen en Henegouwen, P.M.P. Antibody or Antibody Fragments: Implications for Molecular Imaging and Targeted Therapy of Solid Tumors. Front. Immunol. 2017, 8, 1287.
- Quarta, A.; Manna, L.; Pellegrino, T. Antibody-Functionalized Inorganic NPs: Mimicking Nature for Targeted Diagnosis and Therapy. In Bioinspired Approaches for Human-Centric Technologies; Springer International Publishing: New York, NY, USA, 2014; pp. 1–28.
- Oliveira, J.P.; Prado, A.R.; Keijok, W.J.; Antunes, P.W.P.; Yapuchura, E.R.; Guimarães, M.C.C. Impact of Conjugation Strategies for Targeting of Antibodies in Gold Nanoparticles for Ultrasensitive Detection of 17β-Estradiol. Sci. Rep. 2019, 9, 1–8.
- I. N. Peca; A. C. A. Roque; Antibody-Conjugated Nanoparticles for Therapeutic Applications. Current Medicinal Chemistry 2012, 19, 3103-3127, 10.2174/092986712800784667.
- Liébana, S.; Drago, G.A. Bioconjugation and Stabilisation of Biomolecules in Biosensors. Essays Biochem. 2016, 60, 59–68.
- Tallawi, M.; Rosellini, E.; Barbani, N.; Grazia Cascone, M.; Rai, R.; Saint-Pierre, G.; Boccaccini, A.R. Strategies for the Chemical and Biological Functionalization of Scaffolds for Cardiac Tissue Engineering: A Review. J. R. Soc. Interface 2015, 12, 20150254.
- Goossens, J.; Sein, H.; Lu, S.; Radwanska, M.; Muyldermans, S.; Sterckx, Y.G.J.; Magez, S. Functionalization of Gold Nanoparticles with Nanobodies through Physical Adsorption. Anal. Methods 2017, 9, 3430–3440.
- Marques, A.C.; Costa, P.J.; Velho, S.; Amaral, M.H. Functionalizing Nanoparticles with Cancer-Targeting Antibodies: A Comparison of Strategies. J. Control. Release 2020, 320, 180–200.
- Choi, J.S.; Jang, W.S.; Park, J.S. Comparison of Adsorption and Conjugation of Herceptin on Poly(Lactic--Glycolic Acid) Nanoparticles–Effect on Cell Internalization in Breast Cancer Cells. Mater. Sci. Eng. C 2018, 92, 496–507.
- Parracino, M.A.; Martín, B.; Grazú, V. State-of-the-Art Strategies for the Biofunctionalization of Photoactive Inorganic Nanoparticles for Nanomedicine. In Photoactive Inorganic Nanoparticles: Surface Composition and Nanosystem Functionality; Elsevier: Amsterdam, The Netherlands, 2019; pp. 211–257.
- Sivaram, A.J.; Wardiana, A.; Howard, C.B.; Mahler, S.M.; Thurecht, K.J. Recent Advances in the Generation of Antibody-Nanomaterial Conjugates. Adv. Healthc. Mater. 2018, 7, 1700607.
- Polo, E.; Puertas, S.; Moros, M.; Batalla, P.; Guisán, J.M.; De La Fuente, J.M.; Grazú, V. Tips for the Functionalization of Nanoparticles with Antibodies. Methods Mol. Biol. 2013, 1051, 149–163.
- Fager, C.; Olsson, E. Understanding and Utilizing the Biomolecule/Nanosystems Interface: Soft Materials and Coatings for Controlled Drug Release. In Nanotechnologies in Preventive and Regenerative Medicine: An Emerging Big Picture; Elsevier Inc.: Amsterdam, The Netherlands, 2017; pp. 244–260.
- Saha, B.; Songe, P.; Evers, T.H.; Prins, M.W.J. The Influence of Covalent Immobilization Conditions on Antibody Accessibility on Nanoparticles. Analyst 2017, 142, 4247–4256.
- Yao, V.J.; D’Angelo, S.; Butler, K.S.; Theron, C.; Smith, T.L.; Marchiò, S.; Gelovani, J.G.; Sidman, R.L.; Dobroff, A.S.; Brinker, C.J.; et al. Ligand-Targeted Theranostic Nanomedicines against Cancer. J. Control. Release 2016, 240, 267–286.
- Conde, J.; Dias, J.T.; Grazú, V.; Moros, M.; Baptista, P.V.; de la Fuente, J.M. Revisiting 30 Years of Biofunctionalization and Surface Chemistry of Inorganic Nanoparticles for Nanomedicine. Front. Chem. 2014, 2, 48.
- Shen, M.; Rusling, J.F.; Dixit, C.K. Site-Selective Orientated Immobilization of Antibodies and Conjugates for Immunodiagnostics Development. Methods 2017, 116, 95–111.
- Yang, J.; Lee, C.H.; Park, J.; Seo, S.; Lim, E.K.; Song, Y.J.; Suh, J.S.; Yoon, H.G.; Huh, Y.M.; Haam, S. Antibody Conjugated Magnetic PLGA Nanoparticles for Diagnosis and Treatment of Breast Cancer. J. Mater. Chem. 2007, 17, 2695–2699.
- Thermo Fisher Scientific-ES. Sulfhydryl-Reactive Crosslinker Chemistry. Available online: https://www.thermofisher.com/es/es/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/sulfhydryl-reactive-crosslinker-chemistry.html (accessed on 19 August 2020).
- Kantner, T.; Watts, A.G. Characterization of Reactions between Water-Soluble Trialkylphosphines and Thiol Alkylating Reagents: Implications for Protein-Conjugation Reactions. Bioconjug. Chem. 2016, 27, 2400–2406.
- Kharkar, P.M.; Rehmann, M.S.; Skeens, K.M.; Maverakis, E.; Kloxin, A.M. Thiol-Ene Click Hydrogels for Therapeutic Delivery. ACS Biomater. Sci. Eng. 2016, 2, 165–179.
- Agarwal, P.; Bertozzi, C.R. Site-Specific Antibody-Drug Conjugates: The Nexus of Bioorthogonal Chemistry, Protein Engineering, and Drug Development. Bioconjug. Chem. 2015, 26, 176–192.
- Yu, M.K.; Park, J.; Jon, S. Targeting Strategies for Multifunctional Nanoparticles in Cancer Imaging and Therapy. Theranostics 2012, 2, 3–44.
- Akkapeddi, P.; Azizi, S.A.; Freedy, A.M.; Cal, P.M.S.D.; Gois, P.M.P.; Bernardes, G.J.L. Construction of Homogeneous Antibody-Drug Conjugates Using Site-Selective Protein Chemistry. Chem. Sci. 2016, 7, 2954–2963.
- Renault, K.; Fredy, J.W.; Renard, P.Y.; Sabot, C. Covalent Modification of Biomolecules through Maleimide-Based Labeling Strategies. Bioconjugate Chem. 2018, 29, 2497–2513. [Google Scholar] [CrossRef]
- Kim, D.; Herr, A.E. Protein Immobilization Techniques for Microfluidic Assays. Biomicrofluidics 2013, 7, 041501.
- Smith, M.E.B.; Caspersen, M.B.; Robinson, E.; Morais, M.; Maruani, A.; Nunes, J.P.M.; Nicholls, K.; Saxton, M.J.; Caddick, S.; Baker, J.R.; et al. A Platform for Efficient, Thiol-Stable Conjugation to Albumin’s Native Single Accessible Cysteine. Org. Biomol. Chem. 2015, 13, 7946–7949.
- Azadbakht, B.; Afarideh, H.; Ghannadi-Maragheh, M.; Bahrami-Samani, A.; Asgari, M. Preparation and Evaluation of APTES-PEG Coated Iron Oxide Nanoparticles Conjugated to Rhenium-188 Labeled Rituximab. Nucl. Med. Biol. 2017, 48, 26–30.
- Dovgan, I.; Kolodych, S.; Koniev, O.; Wagner, A. 2-(Maleimidomethyl)-1,3-Dioxanes (MD): A Serum-Stable Self-Hydrolysable Hydrophilic Alternative to Classical Maleimide Conjugation. Sci. Rep. 2016, 6, 1–6.
- Markwalter, C.F.; Kantor, A.G.; Moore, C.P.; Richardson, K.A.; Wright, D.W. Inorganic Complexes and Metal-Based Nanomaterials for Infectious Disease Diagnostics. Chem. Rev. 2019, 119, 1456–1518.
- Koniev, O.; Wagner, A. Developments and Recent Advancements in the Field of Endogenous Amino Acid Selective Bond Forming Reactions for Bioconjugation. Chem. Soc. Rev. 2015, 44, 5495–5551.
- Gu, Y.; Zhang, T.T.; Huang, Z.F.; Hu, S.W.; Zhao, W.; Xu, J.J.; Chen, H.Y. An Exploration of Nucleic Acid Liquid Biopsy Using a Glucose Meter. Chem. Sci. 2018, 9, 3517–3522.
- Di Marco, M.; Shamsuddin, S.; Razak, K.A.; Aziz, A.A.; Devaux, C.; Borghi, E.; Levy, L.; Sadun, C. Overview of the Main Methods Used to Combine Proteins with Nanosystems: Absorption, Bioconjugation, and Encapsulation. Int. J. Nanomed. 2010, 5, 37–49.
- Yousefpour, P.; Atyabi, F.; Vasheghani-Farahani, E.; Movahedi, A.A.M.; Dinarvand, R. Targeted Delivery of Doxorubicin-Utilizing Chitosan Nanoparticles Surface-Functionalized with Anti-Her2 Trastuzumab. Int. J. Nanomed. 2011, 6, 1977–1990.
- Eloy, J.O.; Petrilli, R.; Trevizan, L.N.F.; Chorilli, M. Immunoliposomes: A Review on Functionalization Strategies and Targets for Drug Delivery. Colloids Surf. B Biointerfaces 2017, 159, 454–467.
- Yu, K.; Zhou, Y.; Li, Y.; Sun, X.; Sun, F.; Wang, X.; Mu, H.; Li, J.; Liu, X.; Teng, L.; et al. Comparison of Three Different Conjugation Strategies in the Construction of Herceptin-Bearing Paclitaxel-Loaded Nanoparticles. Biomater. Sci. 2016, 4, 1219.
- Chen, Y.; Xianyu, Y.; Wu, J.; Yin, B.; Jiang, X. Click Chemistry-Mediated Nanosensors for Biochemical Assays. Theranostics 2016, 6, 969–985.
- Hein, C.D.; Liu, X.M.; Wang, D. Click Chemistry, a Powerful Tool for Pharmaceutical Sciences. Pharm. Res. 2008, 25, 2216–2230.
- Takayama, Y.; Kusamori, K.; Nishikawa, M. Click Chemistry as a Tool for Cell Engineering and Drug Delivery. Molecules 2019, 24, 172.
- Presolski, S.I.; Hong, V.P.; Finn, M.G. Copper-Catalyzed Azide–Alkyne Click Chemistry for Bioconjugation. Curr. Protoc. Chem. Biol. 2011, 3, 153–162.
- Baskin, J.M.; Prescher, J.A.; Laughlin, S.T.; Agard, N.J.; Chang, P.V.; Miller, I.A.; Lo, A.; Codelli, J.A.; Bertozzi, C.R. Copper-Free Click Chemistry for Dynamic in Vivo Imaging. Proc. Natl. Acad. Sci. USA 2007, 104, 16793–16797.
- Knall, A.C.; Slugovc, C. Inverse Electron Demand Diels-Alder (IEDDA)-Initiated Conjugation: A (High) Potential Click Chemistry Scheme. Chem. Soc. Rev. 2013, 42, 5131–5142.
- Schilling, C.I.; Jung, N.; Biskup, M.; Schepers, U.; Bräse, S. Bioconjugation via Azide-Staudinger Ligation: An Overview. Chem. Soc. Rev. 2011, 40, 4840–4871.
- Liu, Y.; Hou, W.; Sun, H.; Cui, C.; Zhang, L.; Jiang, Y.; Wu, Y.; Wang, Y.; Li, J.; Sumerlin, B.S.; et al. Thiol-Ene Click Chemistry: A Biocompatible Way for Orthogonal Bioconjugation of Colloidal Nanoparticles. Chem. Sci. 2017, 8, 6182–6187.
- Spicer, C.D.; Davis, B.G. Selective Chemical Protein Modification. Nat. Commun. 2014, 5, 1–14.
- Pickens, C.J.; Johnson, S.N.; Pressnall, M.M.; Leon, M.A.; Berkland, C.J. Practical Considerations, Challenges, and Limitations of Bioconjugation via Azide-Alkyne Cycloaddition. Bioconjugate Chem. 2018, 29, 686–701.
- Smyth, T.; Petrova, K.; Payton, N.M.; Persaud, I.; Redzic, J.S.; Graner, M.W.; Smith-Jones, P.; Anchordoquy, T.J. Surface Functionalization of Exosomes Using Click Chemistry. Bioconjug. Chem. 2014, 25, 1777–1784.
- Haldón, E.; Nicasio, M.C.; Pérez, P.J. Copper-Catalysed Azide-Alkyne Cycloadditions (CuAAC): An Update. Org. Biomol. Chem. 2015, 13, 9528–9550.
- Sun, L.; Gai, Y.; Anderson, C.J.; Zeng, D. Highly-Efficient and Versatile Fluorous-Tagged Cu (i)-Catalyzed Azide-Alkyne Cycloaddition Ligand for Preparing Bioconjugates. Chem. Commun. 2015, 51, 17072–17075.
- Zhou, Z.; Badkas, A.; Stevenson, M.; Lee, J.Y.; Leung, Y.K. Herceptin Conjugated PLGA-PHis-PEG PH Sensitive Nanoparticles for Targeted and Controlled Drug Delivery. Int. J. Pharm. 2015, 487, 81–90.
- Badkas, A.; Frank, E.; Zhou, Z.; Jafari, M.; Chandra, H.; Sriram, V.; Lee, J.-Y.; Yadav, J.S. Modulation of in Vitro Phagocytic Uptake and Immunogenicity Potential of Modified Herceptin®-Conjugated PLGA-PEG Nanoparticles for Drug Delivery. Colloids Surf. B Biointerfaces 2018, 162, 271–278.
- Hatit, M.Z.C.; Reichenbach, L.F.; Tobin, J.M.; Vilela, F.; Burley, G.A.; Watson, A.J.B. A Flow Platform for Degradation-Free CuAAC Bioconjugation. Nat. Commun. 2018, 9, 1–7.
- Ramil, C.P.; Lin, Q. Bioorthogonal Chemistry: Strategies and Recent Developments. Chem. Commun. 2013, 49, 11007–11022.
- Agard, N.J.; Prescher, J.A.; Bertozzi, C.R. A Strain-Promoted [3 + 2] Azide-Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems. J. Am. Chem. Soc. 2004, 126, 15046–15047.
- Dommerholt, J.; Rutjes, F.P.J.T.; van Delft, F.L. Strain-Promoted 1, 3-Dipolar Cycloaddition of Cycloalkynes and Organic Azides. In Topics in Current Chemistry; Springer International Publishing: New York, NY, USA, 2016; pp. 1–20.
- Shi, M.; Wosnick, J.H.; Ho, K.; Keating, A.; Shoichet, M.S. Immuno-Polymeric Nanoparticles by Diels-Alder Chemistry. Angew. Chem.-Int. Ed. 2007, 46, 6126–6131.
- Michelle K. Greene; Daniel A. Richards; João C. F. Nogueira; Katrina Campbell; Peter Smyth; Marcos Fernández; Christopher J. Scott; Vijay Chudasama; Forming next-generation antibody–nanoparticle conjugates through the oriented installation of non-engineered antibody fragments. Chemical Science 2018, 9, 79-87, 10.1039/c7sc02747h.
- Gascón, V.; Márquez-Alvarez, C.; Díaz, I.; Blanco, R.M. Hybrid Ordered Mesoporous Materials as Supports for Permanent Enzyme Immobilization through Non-Covalent Interactions. In Non-covalent Interactions in the Synthesis and Design of New Compounds; Wiley: Hoboken, NJ, USA, 2016; pp. 345–360.
- Huang, H.; Oizumi, S.; Kojima, N.; Niino, T.; Sakai, Y. Avidin-Biotin Binding-Based Cell Seeding and Perfusion Culture of Liver-Derived Cells in a Porous Scaffold with a Three-Dimensional Interconnected Flow-Channel Network. Biomaterials 2007, 28, 3815–3823.
- Vadlapudi, A.D.; Vadlapatla, R.K.; Pal, D.; Mitra, A.K. Functional and Molecular Aspects of Biotin Uptake via SMVT in Human Corneal Epithelial (HCEC) and Retinal Pigment Epithelial (D407) Cells. AAPS J. 2012, 14, 832–842.
- Ojima, I.; Wang, X.; Jing, Y.; Wang, C. Quest for Efficacious Next-Generation Taxoid Anticancer Agents and Their Tumor-Targeted Delivery. J. Nat. Prod. 2018, 81, 703–721.
- Chen, S.; Zhao, X.; Chen, J.; Chen, J.; Kuznetsova, L.; Wong, S.S.; Ojima, I. Mechanism-Based Tumor-Targeting Drug Delivery System. Validation of Efficient Vitamin Receptor-Mediated Endocytosis and Drug Release. Bioconjug. Chem. 2010, 21, 979–987.
- Lv, L.; Liu, C.; Chen, C.; Yu, X.; Chen, G.; Shi, Y.; Qin, F.; Ou, J.; Qiu, K.; Li, G. Quercetin and Doxorubicin Co-Encapsulated Biotin Receptortargeting Nanoparticles for Minimizing Drug Resistance in Breast Cancer. Oncotarget 2016, 7, 32184–32199.
- Mehdizadeh, M.; Rouhani, H.; Sepehri, N.; Varshochian, R.; Ghahremani, M.H.; Amini, M.; Gharghabi, M.; Ostad, S.N.; Atyabi, F.; Baharian, A.; et al. Biotin Decorated PLGA Nanoparticles Containing SN-38 Designed for Cancer Therapy. Artif. Cells Nanomed. Biotechnol. 2017, 45, 495–504.
- Krkavcová, E.; Kreisinger, J.; Hyánková, L.; Hyršl, P.; Javůrková, V. The Hidden Function of Egg White Antimicrobials: Egg Weightdependent Effects of Avidin on Avian Embryo Survival and Hatchling Phenotype. Biol. Open 2018, 7, 031518.
- Jain, A.; Barve, A.; Zhao, Z.; Jin, W.; Cheng, K. Comparison of Avidin, Neutravidin, and Streptavidin as Nanocarriers for Efficient SiRNA Delivery. Mol. Pharm. 2017, 14, 1517–1527.
- Verdoliva, A.; Bellofiore, P.; Rivieccio, V.; Catello, S.; Colombo, M.; Albertoni, C.; Rosi, A.; Leoni, B.; Anastasi, A.M.; De Santis, R. Biochemical and Biological Characterization of a New Oxidized Avidin with Enhanced Tissue Binding Properties. J. Biol. Chem. 2010, 285, 9090–9099.
- Fahie, M.A.; Chen, M. Electrostatic Interactions between OmpG Nanopore and Analyte Protein Surface Can Distinguish between Glycosylated Isoforms. J. Phys. Chem. B 2015, 119, 10198–10206.
- Jain, A.; Cheng, K. The Principles and Applications of Avidin-Based Nanoparticles in Drug Delivery and Diagnosis. J. Control. Release 2017, 245, 27–40.
- Sperling, R.A.; Parak, W.J. Surface Modification, Functionalization and Bioconjugation of Colloidal Inorganic Nanoparticles. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2010, 368, 1333–1383.
- Müller, J.M.; Risse, J.M.; Jussen, D.; Flaschel, E. Development of Fed-Batch Strategies for the Production of Streptavidin by Streptomyces Avidinii Based on Power Input and Oxygen Supply Studies. J. Biotechnol. 2013, 163, 325–332.
- Wu, S.C.; Wang, C.; Hansen, D.; Wong, S.L. A Simple Approach for Preparation of Affinity Matrices: Simultaneous Purification and Reversible Immobilization of a Streptavidin Mutein to Agarose Matrix. Sci. Rep. 2017, 7, 1–10.
- Nguyen, T.T.; Sly, K.L.; Conboy, J.C. Comparison of the Energetics of Avidin, Streptavidin, NeutrAvidin, and Anti-Biotin Antibody Binding to Biotinylated Lipid Bilayer Examined by Second-Harmonic Generation. Anal. Chem. 2012, 84, 201–208.
- Kroetsch, A.; Chin, B.; Nguyen, V.; Gao, J.; Park, S. Functional Expression of Monomeric Streptavidin and Fusion Proteins in Escherichia Coli: Applications in Flow Cytometry and ELISA. Appl. Microbiol. Biotechnol. 2018, 102, 10079–10089.
- Bigini, P.; Previdi, S.; Casarin, E.; Silvestri, D.; Violatto, M.B.; Facchin, S.; Sitia, L.; Rosato, A.; Zuccolotto, G.; Realdon, N.; et al. In Vivo Fate of Avidin-Nucleic Acid Nanoassemblies as Multifunctional Diagnostic Tools. ACS Nano 2014, 8, 175–187.
- Roncato, F.; Rruga, F.; Porcù, E.; Casarin, E.; Ronca, R.; Maccarinelli, F.; Realdon, N.; Basso, G.; Alon, R.; Viola, G.; et al. Improvement and Extension of Anti-EGFR Targeting in Breast Cancer Therapy by Integration with the Avidin-Nucleic-Acid-Nano-Assemblies. Nat. Commun. 2018, 9, 1–11.
- Elzahhar, P.; Belal, A.S.F.; Elamrawy, F.; Helal, N.A.; Nounou, M.I. Bioconjugation in Drug Delivery: Practical Perspectives and Future Perceptions. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2019; Volume 2000, pp. 125–182.
- Ren, W.X.; Han, J.; Uhm, S.; Jang, Y.J.; Kang, C.; Kim, J.H.; Kim, J.S. Recent Development of Biotin Conjugation in Biological Imaging, Sensing, and Target Delivery. Chem. Commun. 2015, 51, 10403–10418.
- Wartlick, H.; Michaelis, K.; Balthasar, S.; Strebhardt, K.; Langer, K. Highly Specific HER2-Mediated Cellular Uptake of Antibody-Modified Nanoparticles in Tumour Cells. J. Drug Target. 2004, 12, 461–471.
- Powell, D.; Chandra, S.; Dodson, K.; Shaheen, F.; Wiltz, K.; Ireland, S.; Syed, M.; Dash, S.; Wiese, T.; Mandal, T.; et al. Aptamer-Functionalized Hybrid Nanoparticle for the Treatment of Breast Cancer. Eur. J. Pharm. Biopharm. 2017, 114, 108–118.
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against her2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792.


